Location: Home >> Detail
Immunometabolism. 2020;2(2):e200010. https://doi.org/10.20900/immunometab20200010

Department of Surgery, Children’s Hospital of Pittsburgh, University of Pittsburgh Medical Center, Pittsburgh, PA 15260, USA
* Correspondence: Jon D. Piganelli, Tel.: +1-412-692-7498.
This article belongs to the Virtual Special Issue "Immunometabolism in Autoimmune Diseases"
Advances in the field of immunometabolism have indicated that cellular metabolic programs control T cell differentiation and effector functions. As CD4+ T cells are significant contributors to destruction of the insulin-secreting β cell in Type 1 Diabetes (T1D), strategies to limit autoreactive T cell activation and promote durable tolerance have been explored in an effort to prevent or delay disease onset. Interestingly, substantial evidence indicates that autoreactive T cells are at a heightened activation status associated with dysregulation of metabolic pathways, with distinct nutrient preferences and needs of their own. While largely unexplored in T1D, specific metabolic pathways have been exploited to correct known defects in several autoimmune diseases including but not limited to Systemic Lupus Erythematosus (SLE), Multiple Sclerosis (MS), and Rheumatoid Arthritis (RA). These therapeutic strategies have been focused on targeting glycolysis, which is the major pathway used by activated T cells as it is required for acquisition of effector functions. Use of glycolysis inhibitors such as 2-Deoxy-D-glucose (2-DG), have shown impressive success in limiting T cell mediated damage that could prevent or reverse onset of autoimmunity. More importantly, while specificity has been a major hurdle for use of immunotherapies, regulating immunity by targeting metabolism resulted in selective targeting of autoreactive T cells while leaving global immune function intact, a phenomenon commonly referred to as “cellular selectivity based on demand”. This review will outline examples of metabolic regulation success in autoimmunity and provide evidence to support use of these inhibitors in T1D, where new therapeutic strategies are desperately needed.
2-DG, 2-Deoxy-D-glucose; AKT, protein kinase B; AMPK, AMP-activated protein kinase; APC, antigen presenting cell; ASCT2, alanine-serine-cysteine transporter 2; ATP, adenosine triphosphate; Bcl-2, B-cell lymphoma 2; CPT1α, carnitine palmitoyltransferase Iα; CTL, cytotoxic T lymphocyte; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; DC, dendritic cell; EAE, experimental autoimmune encephalomyelitis; ECAR, extracellular acidification rate; ER, endoplasmic reticulum; Electron Transport Chain, ETC; fatty acid oxidation, FAO; FLS, fibroblast-like synoviocyte; FoxP3, forkhead box P3; Glut1, glucose transporter 1; HIF1α, hypoxia-inducible factor 1α; HLA, human leukocyte antigen; interferon-γ, IFNγ; interleukin-2, IL-2; interleukin-1β, IL-1β; lymphocyte activation gene-3, Lag-3; MHC-II, major histocompatibility complex class II; MHP, mitochondrial inner-membrane hyperpolarization; MnP, manganese metalloporphyrin; MS, multiple sclerosis; mTORC1, mammalian target of rapamycin complex I; mTOR, mammalian target of rapamycin; Myc, myelocytomatosis oncogene; NAC, N-acetylcysteine; NADH, nicotinamide adenine dinucleotide; NOD, non-obese diabetic mouse; OCR, oxygen consumption rate; oxidative phosphorylation, OXPHOS; PBMC, peripheral blood mononuclear cells; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; RA, rheumatoid arthritis; ROS, reactive oxygen species; SLE, systemic lupus erythematosus; STAT3, signal transducer and activator of transcription 3; T-bet, T-box protein; T1D, type 1 diabetes; T2D, type 2 diabetes; TCR, T cell receptor; Tfh, follicular T helper cells; TIL, tumor-infiltrating lymphocyte; TIM3, T cell immunoglobulin mucin domain-3; TME, tumor microenvironment; TNFα, tumor necrosis factor α; Treg, regulatory T cell; WT, wild type
T1D is an autoimmune disease in which pancreatic islet β cells are targeted and destroyed by the immune system [1]. 1.25 million Americans are living with T1D, including 200,000 youth and over a million adults [2]. If current trends continue, 5 million people in the U.S. are expected to have T1D by 2050, including 600,000 people under the age of 20 years old [2]. Since the 1920’s, patients with T1D have been treated by the daily administration of exogenous insulin in order to maintain euglycemia; however, insulin is not a cure for these individuals. Complications still arise from exogenous insulin use, as a number of patients experience low blood sugar unawareness and increased susceptibility to infections, further impacting their day-to-day activities [1–4]. As a result, less than one-third of people with T1D are achieving target blood glucose levels. Furthermore, while the life expectancy of patients with T1D continues to improve, it remains reduced by approximately 20 years compared to the general population [1–4]. For these reasons, new and innovative preventative measures are needed to combat T1D.
As is the case in several autoimmune diseases, the activation of autoreactive CD4+ T cells that escape central tolerance and bypass tolerogenic mechanisms in the periphery drive T1D pathogenesis. Therefore, understanding the mechanisms governing T cell activation and aberrant immune-mediated responses are vital for finding druggable therapeutic targets for disease prevention or treatment. An emerging concept in immunology is that metabolic reprogramming and lymphocyte activation are intricately linked, as both T cell fate and function are dependent on cellular metabolism [5–13]. Consequently, cellular metabolism has the ability to influence the final outcome of the innate and adaptive immune responses, further impacting health and disease outcome [8]. Studies interrogating T cell activation and function in animal models and human patients with T1D have indicated autoreactive T cells may have a heightened activation status [14–16]. This altered status, along with functionally defective Regulatory T cells (Tregs), leads to T cell activation, enhanced effector capabilities, and excessive proinflammatory cytokine secretion, especially interferon-γ (IFNγ). Based on these observations and due to the importance of metabolism in dictating T cell functions, it is clear that autoreactive T cells do not play by the same rules metabolically compared to their healthy counterparts. Indeed, self-reactive T cells appear to have a multitude of metabolic irregularities that contribute to their ability to cause and mediate disease progression [14–16]. This metabolic dysregulation has opened the door for targeting metabolism to regulate T cell responses and restore tolerance to self-antigens in autoimmunity, however whether metabolic modulation holds the future of T1D therapeutics remains unknown. Herein, this review will highlight evidence that enhanced cellular bioenergetics plays a role in how self-reactive T cells function; provide examples on how manipulating T cell metabolism has been used therapeutically to prevent and reverse autoimmune pathologies, and discuss opportunities for metabolic intervention to be used to selectively target β cell reactive CD4+ T cells in T1D, which up to this point has been an untapped opportunity.
Invasion of the pancreatic islets by immune cells (insulitis) is a hallmark of T1D pathogenesis that leads to progressive destruction of the β cell and precedes disease onset. This along with a break in tolerance to β cell antigens lead to the activation and clonal expansion of autoreactive CD4+ T cells that drive T1D. While the etiology of T1D is not completely understood, we do know that in those with a genetic predisposition to autoimmunity, a triggering event (i.e., chemical exposure, dysglycemia, endoplasmic reticulum (ER) stress, reactive oxygen species (ROS), or viral infection) affecting β cells in the pancreas leads to release of β cell antigens that are phagocytosed by antigen presenting cells (APCs), like macrophages and dendritic cells (DCs), and leads to their subsequent activation [17–22]. Activated APCs traffic to the pancreatic draining lymph node where they present β cell self-antigens to autoreactive CD4+ T cells that become activated and travel back to the pancreas as effector CD4+ T cells. Effector CD4+ T cells coordinate the resulting autoimmune attack of the β cell via secretion of proinflammatory cytokines like IFNγ and tumor necrosis factor-α (TNFα) that can directly cause β cell death, provide help to activate CD8+ T cells and B cells, and provide an inflammatory milieu that favors continual APC activation [17,20]. Ultimately, this perpetuates a cycle of autoimmune attack that when left unhindered, leads to the organ-specific autoimmunity and β cell destruction that culminates in hyperglycemia and T1D onset (Figure 1) [17,20]. Orchestration of autoimmune responses by APCs, T cells, and B cells together leads to β cell death and hyperglycemia. The diversity of immune cells involved in autoimmune attack of the β cell provide a variety of potential therapeutic targets to control aberrant responses in T1D. Further, finding ways to alter the differentiation of immune cell subsets to more anti-inflammatory phenotypes or impeding activation of autoreactive T cells will allow for the design of novel therapies to prevent, delay, or reverse T1D.
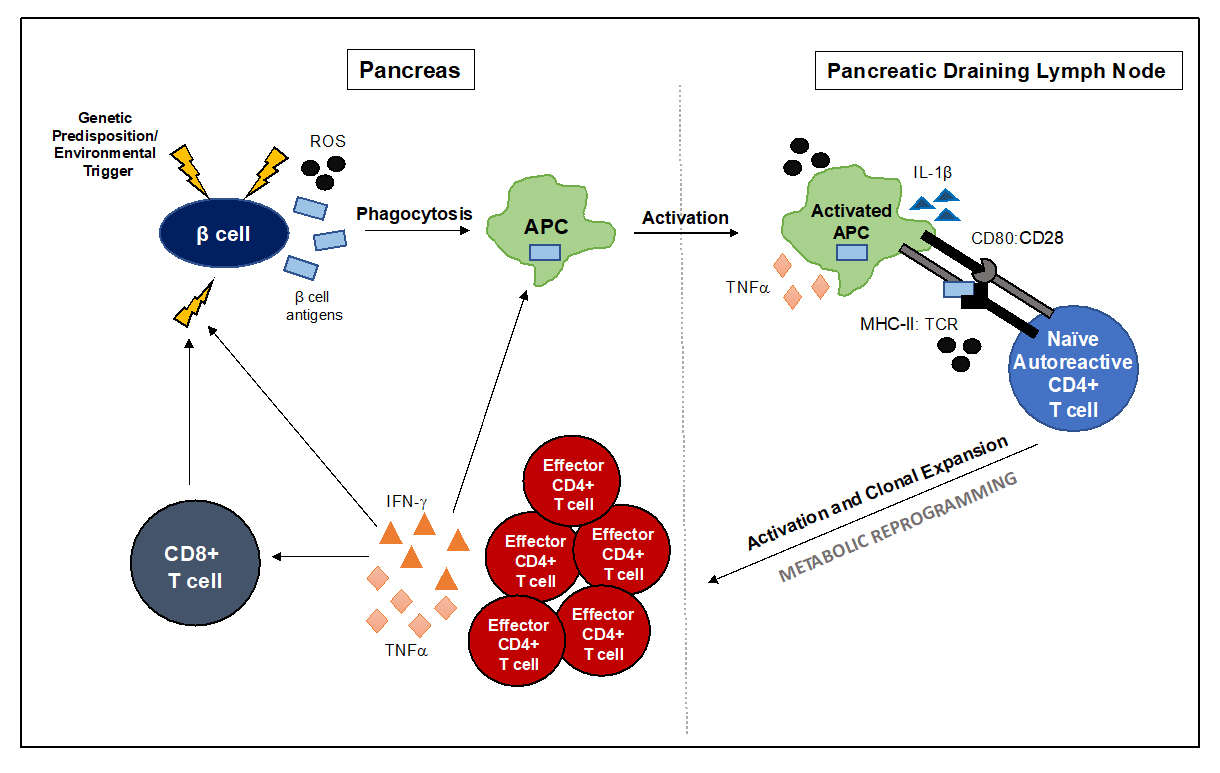 Figure 1. Immune-mediated β cell killing leads to onset of T1D. In genetically predisposed individuals, an initial threat targeting the β cell triggers the release of β cell antigens in the pancreas. These autoantigens are phagocytosed and processed by islet-resident macrophages and DC that become activated and traffic to the pancreatic draining lymph node. It is hear that naïve autoreactive CD4+ T cells specific for islet antigens become educated, leading to their subsequent activation and clonal expansion, supported in large part by dramatic changes in cellular metabolism. Once activated, effector CD4+ T cells travel back to the target organ where they are instrumental in orchestrating autoimmune attack of the β cell that leads to β cell death and hyperglycemia.
Figure 1. Immune-mediated β cell killing leads to onset of T1D. In genetically predisposed individuals, an initial threat targeting the β cell triggers the release of β cell antigens in the pancreas. These autoantigens are phagocytosed and processed by islet-resident macrophages and DC that become activated and traffic to the pancreatic draining lymph node. It is hear that naïve autoreactive CD4+ T cells specific for islet antigens become educated, leading to their subsequent activation and clonal expansion, supported in large part by dramatic changes in cellular metabolism. Once activated, effector CD4+ T cells travel back to the target organ where they are instrumental in orchestrating autoimmune attack of the β cell that leads to β cell death and hyperglycemia.
While a number of immune cells are involved in autoimmune destruction of the β cell, autoreactive CD4+ T cells are largely responsible for mediating and driving disease pathogenesis [1,4,17,18,20–28]. This is also true for other autoimmune diseases like SLE, MS, and RA. CD4+ T cells are fundamental and required for T1D development, as studies in the non- obese diabetic (NOD) mouse have shown that depletion of CD4+ T cells or treatment with non-depleting anti-CD4 antibodies prevents disease onset [23,29–31]. Further, genetic predisposition to autoimmunity and T1D disease development has been linked to human leukocyte antigen (HLA) class II alleles DR4, DQ8, and DQ2, which confer the highest genetic risk of T1D in human patients. This suggests that HLA class II restricted CD4+ T cells play a key role in T1D [23,32]. While CD8+ T cell mediated β cell killing is a major mechanism of disease development, CD4+ T cells lead the way in orchestrating the attack against islet β cells [21,23,24]. While their role includes providing help to activate CD8+ T cells, B cells, and antigen presenting cells, activation of CD4+ T cells and their subsequent secretion of proinflammatory cytokines, especially IFNγ, TNFα, and interleukin-1β (IL-1β), can directly induce β cell apoptosis [1,17,21,23,24]. Without intervention, this results in a cycle of β cell killing, resulting in dysregulation of dynamic glucose sensing and subsequent insulin secretion [17]. Therefore, understanding the mechanisms governing CD4+ T cell effector function, and using this knowledge to find therapeutic targets to inhibit autoreactive T cell activation are of the utmost importance, as they are critical for disease prevention and future therapies.
The ability of autoreactive T cell clones to escape deletion during development in the thymus leads to autoimmunity [21,22,26,28,33]. However, a failure in peripheral tolerance mechanisms to control these rogue T cells also plays a significant role. Tregs (simply defined as CD4+ CD25+ Forkhead box P3+ (FoxP3; master regulator of Treg development and function)), are members of the CD4 compartment and are widely regarded as the primary mediators of peripheral tolerance [4,22,28]. However, in T1D and other autoimmune disorders, Tregs are known to be functionally defective. In T1D specifically, Tregs have an altered phenotype, most notably in terms of their stability, survival, and functionality [21–23,26,28,33–36]. While there appears to be no change in levels of circulating Tregs in mice or human patients with T1D, previous studies have shown a decline in intra-islet Treg frequencies that correlated with disease development in the NOD mouse [34,36]. Moreover, a number of investigations have shown that FoxP3+ Treg function and suppressive capacity is defective in those with T1D, as Tregs from affected individuals were less able to control the proliferation of autologous T effectors when compared to healthy controls [22,34–36] Further, Tregs in autoimmunity suffer from lineage instability, as emerging data suggests that functional Tregs can either convert to effector T cells or become Th1-like Treg cells associated with reduced or loss of FoxP3 expression and suppressive function, expression of the Th1 specific transcription factor T-box protein expressed in T cells (T-bet), and enhanced proinflammatory cytokine secretion, especially IFNγ [37,38]. Further insight is needed to discern the causative reason behind impaired Treg stability and function in T1D, as reinvigorating Treg function may promote tolerance in those predisposed to autoimmunity.
Cellular metabolism dictates T cell fate and function. Consequently, cellular metabolism has the ability to influence the final outcome of the innate and adaptive immune responses, further impacting health and disease outcome [8]. T cells undergo substantial metabolic reprogramming from oxidative phosphorylation (OXPHOS) to aerobic glycolysis during activation upon encounter with cognate antigen [6–10,12,13,15,39]. This section will briefly review the major metabolic pathways utilized by T cells during activation and quiescence, however this topic has been reviewed in more detail in reports cited here [7,9,10,12].
As T cells move through the various stages of their life cycle, their metabolic demands vary. Naïve CD4+ T cells are metabolically quiescent, as they utilize OXPHOS via the mitochondria to support basic cellular processes, maintain homeostasis, and to circulate the periphery surveying the body for foreign invaders [7,9,10,13]. In fact, 96% of the adenosine triphosphate (ATP) generated by naïve T cells comes from oxidative metabolism, with roughly 4% coming from glycolytic metabolism [12]. Upon recognition of foreign antigen, along with the necessary costimulatory signals, naïve T cells become activated and undergo metabolic reprogramming to support this change in function. During activation, T cells switch to a program of anabolic growth and biomass accumulation to generate daughter cells, requiring an increased demand for ATP and metabolic resources [7–10,12,13]. This developmental program is characterized by an increase in rapid growth, proliferation, and acquisition of specialized effector functions [7,9,10,13,39]. Activated T cells are more metabolically active, and engage primarily in aerobic glycolysis, marked by the conversion of glucose to lactate despite the presence or availability of oxygen [9,10]. This phenomenon, known as the Warburg Effect, was characterized by Dr. Otto Warburg in the 1920’s, and is a hallmark of cancer metabolism [11,40]. Although far less efficient than OXPHOS in terms of energy production, aerobic glycolysis allows for proliferating T cells to generate ATP quickly, and also allows for the generation of metabolic intermediates important for clonal expansion and effector capabilities [8,10]. After clearance of the initial threat, most effector T cells die, however a small population of long-lived antigen-specific memory T cells remain that mediate protection against re-challenge [41]. Memory T cells, much like naïve T cells, are quiescent and rely primarily on OXPHOS and fatty acid oxidation (FAO) for their development and long-term survival [7,9,10] (Figure 2).
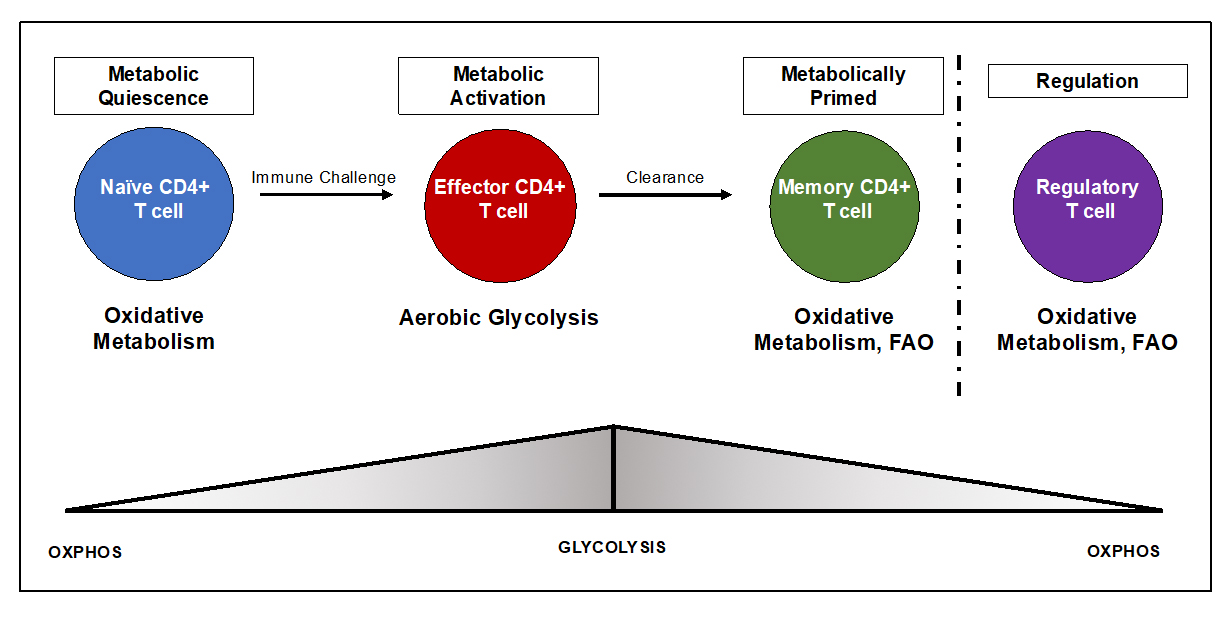 Figure 2. Distinct metabolic profiles dictate T cell fate and function. The metabolic needs of CD4+ T cells drastically change over the course of an immune response. Consequently, immunometabolism has the ability to dictate whether a pro- or anti- inflammatory response will ensue. While there is some layover in the metabolic programs of different T cell subsets, clear distinctions exist. Naïve T cells are quiescent and utilize OXPHOS to generate ATP to support antigenic surveillance. Upon encounter with antigen, naïve T cells become activated and undergo a metabolic to utilize aerobic glycolysis, which is vital for T cell proliferation and acquisition of effector functions. After clearance, a small subset of T cells become memory T cells and revert back to a program of FAO and OXPHOS, which promotes longevity and survival. Like memory T cells, Tregs too are thought to utilize FAO. This pathway has been implicated in supporting Treg stability and suppressive capabilities.
Figure 2. Distinct metabolic profiles dictate T cell fate and function. The metabolic needs of CD4+ T cells drastically change over the course of an immune response. Consequently, immunometabolism has the ability to dictate whether a pro- or anti- inflammatory response will ensue. While there is some layover in the metabolic programs of different T cell subsets, clear distinctions exist. Naïve T cells are quiescent and utilize OXPHOS to generate ATP to support antigenic surveillance. Upon encounter with antigen, naïve T cells become activated and undergo a metabolic to utilize aerobic glycolysis, which is vital for T cell proliferation and acquisition of effector functions. After clearance, a small subset of T cells become memory T cells and revert back to a program of FAO and OXPHOS, which promotes longevity and survival. Like memory T cells, Tregs too are thought to utilize FAO. This pathway has been implicated in supporting Treg stability and suppressive capabilities.
Tregs too have their own metabolic preferences that dictate their function and stability, however there have been conflicting reports in both mouse and human studies, as it appears Tregs utilize a number of metabolic programs depending on their environment and specific needs [5,7,9,10,12,38,39,42–46]. Overall, Tregs are thought to preferentially rely on oxidative metabolism [39,42] (Figure 2). This idea was supported in a paper by Angelin et al., where it was determined that FoxP3 induced OXPHOS and shut down glycolysis via binding to the myelocytomatosis oncogene (Myc) (transcription factor that drives glycolytic metabolism and metabolic reprogramming) gene promoter and suppressing Myc gene expression [47]. More specifically, Tregs rely on FAO, which feeds into the mitochondria to supply OXPHOS, and readily take up externally derived fatty acids to support high rates of FAO. Interestingly, treatment with etomoxir, an inhibitor of the enzyme Carnitine palmitoyltransferase Iα (CPT1α), which converts acyl-CoAs to acylcarnitines allowing for entry of fatty acids into the mitochondria and FAO, abrogates Treg development and suppressive function suggesting that Tregs depend on the FAO pathway to differentiate and perform their effector functions [7,36,39,44,45]. On the other hand, Tanimine et al. and others have indicated that glycolysis controls the induction of functional FoxP3 during early activation and proliferation in Tregs isolated from human subjects [48]. Another study of human Tregs found that the glycolytic enzyme enolase-1 was required for Treg suppressive capacity via control of FoxP3 splice variants [49]. While the exact metabolic preferences of Tregs remain elusive, it’s clear that these cells display a mixed metabolism depending on nutrient availability and environmental cues. Further studies are desperately needed to fully understand the metabolic requirements of Tregs in order to have a better understanding of mechanisms driving Treg differentiation, stability, and functional capacity. All in all, while each phase of the T cell life cycle has distinct metabolic requirements, each program allows for T cells to function and respond to various situations effectively during their life span. Moreover, as cellular metabolism is responsible for shaping immune responses, it’s clear that changes in immunometabolism drive autoimmunity, specifically T1D pathogenesis (Figure 3).
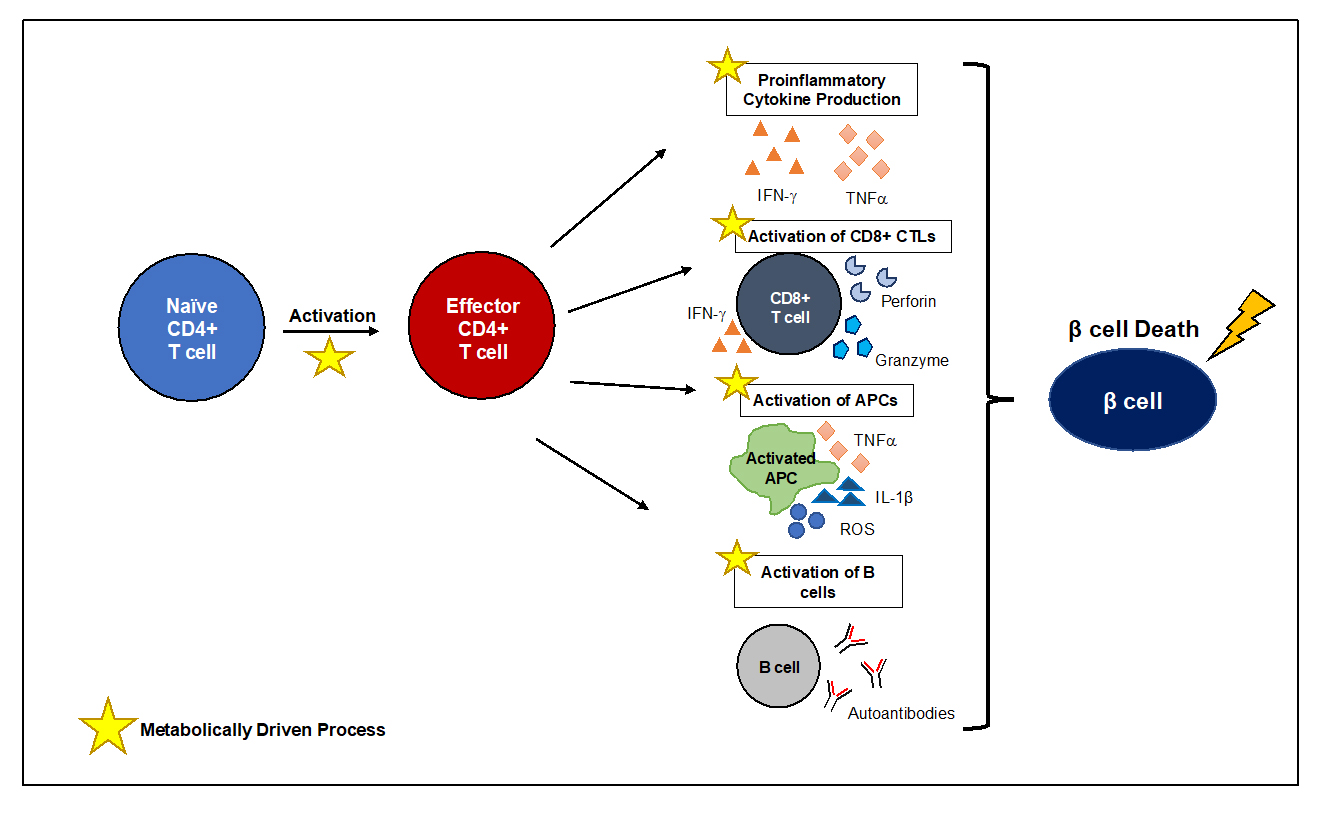 Figure 3. Changes in Immunometabolism drive β cell death in T1D. T1D pathogenesis is largely mediated by the activation of autoreactive CD4+ T cells that is fueled by metabolic reprogramming. Activation of CD4+ T cells is a metabolically demanding process accompanied by a metabolic switch to aerobic glycolysis, that although less efficient, is required for T cell clonal expansion and effector capabilities. Like CD4+ T cells, other immune cells undergo metabolic reprogramming to support immune function. CD4+ T cells are critical mediators of orchestrating β cell destruction, where proinflammatory cytokine production can lead to direct β cell death and support the downstream activation of other immune cells like CD8+ CTLs, APCs, and B cells. The importance of metabolic reprogramming and immune cell activation therefore provide an opportunity to modulate immune cell activation and differentiation by exploiting specific bioenergetic programs to limit autoimmunity.
Figure 3. Changes in Immunometabolism drive β cell death in T1D. T1D pathogenesis is largely mediated by the activation of autoreactive CD4+ T cells that is fueled by metabolic reprogramming. Activation of CD4+ T cells is a metabolically demanding process accompanied by a metabolic switch to aerobic glycolysis, that although less efficient, is required for T cell clonal expansion and effector capabilities. Like CD4+ T cells, other immune cells undergo metabolic reprogramming to support immune function. CD4+ T cells are critical mediators of orchestrating β cell destruction, where proinflammatory cytokine production can lead to direct β cell death and support the downstream activation of other immune cells like CD8+ CTLs, APCs, and B cells. The importance of metabolic reprogramming and immune cell activation therefore provide an opportunity to modulate immune cell activation and differentiation by exploiting specific bioenergetic programs to limit autoimmunity.
Studies investigating the mechanisms that dictate T cell activation and function have shed light on the importance of cellular metabolism in guiding these processes. While there is some layover in terms of the pathways that are vital for immune cell function, metabolic signatures exist between T cell subsets and are important in controlling T cell differentiation [6,7,9,10,39]. Further, different metabolic processes can influence the final outcome of an immune response and can govern whether a pro- or anti- inflammatory response will ensue. In the case of autoimmunity, recent work has highlighted that metabolic differences exist between immune cells in healthy individuals versus in patients with autoimmune disorders [14–16]. Due to the importance of metabolism in controlling cell fate and function, as well as the importance of CD4+ T cells that coordinate autoimmune attack of the β cell in T1D, understanding metabolic signatures of autoreactive CD4+ T cell subsets in autoimmunity are vital to providing new therapeutic targets to control these responses. The following section will review immunometabolism as a therapeutic target to control autoreactive T cell responses in SLE, MS, and RA. This will afford insight on the metabolic requirements of autoreactive T cells and provide evidence to support metabolic regulation as the future of immunotherapies in autoimmunity, especially in T1D.
Systemic Lupus ErythematosusStudies focused specifically on the role of immunometabolism in autoimmunity have indicated that autoreactive T cells display altered metabolic dependencies and programs [14–16]. Interestingly, self-reactive T cells from different autoimmune diseases studied thus far display distinct metabolic programs with little overlap, further indicating a role for the specific immune microenvironment in driving T cell differentiation and effector function. Due to the importance of effector T cells in orchestrating attack of the various tissues targeted in autoimmunity, inhibiting the activation of these CD4+ T cells is an ideal therapeutic target [12,46,50]. Generally speaking, autoreactive T cells are thought to have enhanced bioenergetics compared to healthy controls, although the metabolic demands of autoreactive T cells in T1D specifically remain largely unexplored. This was demonstrated in a lupus model by Yin et al., where it was determined that both glycolysis and mitochondrial oxidative metabolism, as well as mammalian target of rapamycin complex 1 (mTORC1) activity, are elevated in CD4+ T cells from lupus prone mice compared to non-autoimmune control animals [51]. The authors concluded that this metabolic dysregulation was a major contributor to the immunological abnormalities associated with lupus pathogenesis including T cell hyperactivation and increased IFNγ production [51]. The enhanced cellular metabolism of CD4+ T cells from lupus prone animals preceded onset of disease and increased as T cells became more activated throughout disease progression, indicating that metabolic differences may be able to be specifically manipulated to prevent onset of autoimmunity prior to CD4+ T cell hyperactivation [51]. In an effort to normalize autoreactive CD4+ T cell functions in vitro and in vivo, the authors utilized a combination treatment of the glycolysis inhibitor 2-DG and metformin, an inhibitor of mitochondrial metabolism (specifically Complex 1 of the electron transport chain (ETC). In vitro, this treatment combination inhibited excessive IFNγ production and promoted interleukin-2 (IL-2) production which is otherwise defective during lupus [51]. In vivo, targeting glucose and mitochondrial metabolism reversed disease and simultaneously lead to significant decreases in glycolysis and OXPHOS in autoreactive T cells to levels similar to that of non-autoimmune prone controls, decreased the expression of activation and effector function markers, decreased autoantibody production, and improved production of IL-2 by autoreactive CD4+ T cells; ultimately leading to significant reductions in disease pathology [51]. This enhanced bioenergetic profile of lupus-prone CD4+ T cells was also seen in T cells from human SLE patients, and could be normalized via treatment with metformin [52]. While treatment with either 2-DG or metformin alone were sufficient to prevent disease, combination therapy was required for disease reversal. Moreover, a continuous treatment was required to maintain tolerance, as cessation of treatment lead to disease flare-ups [52].
Recent work by Choi et al. confirmed that CD4+ T cells from lupus prone animals were glycolytic, and found that Inhibiting glucose metabolism selectively targeted autoreactive follicular helper T cells (Tfh), which are expanded in SLE and are required for production of high affinity autoantibodies [53]. This treatment was capable of preventing onset of disease in various mouse models of SLE. Use of CG-5, an inhibitor of glucose transporter, was also capable of ameliorating autoimmune activation in lupus [54]. Tfh cells from lupus prone animals displayed increased mTORC1 activation compared to B6 controls, consistent with the findings by Yin et al., and had a pre-activation status indicated by higher mTORC1 activation in naïve CD44-negative CD4+ T cells in lupus-prone animals [51–53]. Moreover, CD4+ T cells from lupus animals displayed increased expression of the pro-survival factor B-cell lymphoma 2 (Bcl2), indicating a resistance of autoreactive T cells to apoptosis. Preventative treatment (animals were autoantibody positive without clinical disease) with 2-DG reduced autoantibody production as well as the frequency and number of Tfh and germinal center B cells and reduced the number of total splenic CD4+ T cells in four lupus-prone mouse strains, but not in control B6 mice [53]. Interestingly, treatment with 2-DG had variable effects on CD4+ T cell activation, as activation and CD44+CD4+ T cell frequency were only decreased in a few of the lupus models tested, indicating that spontaneous lupus Tfh cells are uniformly sensitive to glycolysis inhibition compared to other T cell subsets that showed a variable response depending on strain. Finally, while proliferation and signal transducer and activator of transcription 3 (STAT3) signaling were similar between lupus-prone and B6 Tfh cells, these were increased in naïve TC CD4+ T cells in which glycolysis is also higher than in naïve B6 CD4+ T cells. This finding is interesting, in that it indicates that autoreactive CD4+ T cells display heightened bioenergetics that may contribute to the ability for autoreactive T cells to escape deletion and persist in the periphery. This heightened activation status of naïve CD4+ T cells in autoimmune prone animals also gives precedence for selectively targeting CD4+ T cell metabolism as a means to control autoimmunity [51–53].
As was demonstrated in mouse models of SLE, CD4+ T cells from human patients also exhibit elevated cellular metabolism compared to CD4+ T cells from healthy control patients [51]. Enhanced bioenergetics correlated with increased production of IFNγ that could be normalized in vitro via treatment with Metformin [51]. These data corroborate earlier studies that demonstrated lupus T cells from human patients have increased mTOR activity, mitochondrial dysfunction, increased oxidative stress, and glutathione depletion [55–60]. Aberrant oxidative stress and ROS in SLE T cells was attributed to increased oxygen consumption and mitochondrial ETC complex I activity [56]. As ROS are important mediators in rewiring CD4+ T cell metabolism during activation, the phenotype exhibited by SLE T cells is indicative of enhanced bioenergetics, aberrant T cell activation, and overall T cell dysfunction that contributes to SLE immunopathology [58]. Further, higher mTOR activity leads to expansion of pathogenic Th17 cells and reduction of functional Tregs, further contributing to autoimmunity [57]. Interestingly, treatment with rapamycin or the antioxidant N-acetylcysteine (NAC) were capable of reducing SLE disease activity by decreasing mTOR activity in effector T cells, expanding Tregs, and enhancing Treg suppressive functions [55–57,59–61]. All in all, these studies in murine and human models of SLE disease indicate metabolic aberrations exist in the CD4+ T cell compartment compared to healthy counterparts, and that targeting metabolic pathways can reverse T cell dysfunction to correct known defects that contribute to SLE pathogenesis.
Multiple SclerosisThe metabolic profile of CD4+ T cells from patients with multiple sclerosis (MS) also display altered bioenergetics compared to those of healthy patients, however the literature has had conflicting reports. As is the case in other autoimmune disorders, Th17 cells contribute to autoimmune disease pathogenesis and progression, where a metabolic switch to glycolysis accompanies T cell activation and drives effector functions [9,10,39,62]. This finding that Th17s relied on glycolysis was confirmed in a study by Shi et al., where it was demonstrated that Th17 differentiation was dependent on hypoxia-inducible factor 1α (HIF1α) dependent up-regulation of glycolytic activity, as CD4+ T cells deficient in HIF1α had diminished Th17 differentiation due to a failure to upregulate the glycolytic enzymes required for metabolic reprogramming during activation [46]. Further, blocking glycolysis via 2-DG or the mammalian target of rapamycin (mTOR) inhibitor Rapamycin reduced Th17 differentiation of naïve T cells in vitro and reduced the ability of Th17 to cause experimental autoimmune encephalomyelitis (EAE), a murine model of MS, in vivo [46]. This was associated with reduced lymphocyte infiltration and spinal cord inflammation [46]. Other work in EAE has demonstrated the importance of glutamine in driving autoreactive CD4+ T cell activation, differentiation, and effector functions, and is dependent on the amino acid glutamine transporter alanine-serine-cysteine transporter 2 (ASCT2) [63]. Indeed, genetic knockout of ASCT2 resulted in reduced Th1 and Th17 differentiation, cytokine production, reduced leukocyte infiltration into the central nervous system, and reduced EAE disease severity compared to wild type (WT) controls [63]. Interestingly, the importance of glutamine in mouse models of MS was also true in patients, where increased concentrations of glutamine and glutamate has been found in the cerebrospinal fluid and brain biopsies from affected individuals. Increases in these metabolites were also associated with disease severity and have been suggested to be potential biomarkers for MS [64,65].
In human patients, altered energy metabolism has been a common finding, however the exact metabolic phenotype of autoreactive CD4+ T cells remain elusive. Studies analyzing the gene-expression profile in brains from post-mortem MS subjects have showed evidence of an upregulation of genes involved in cellular metabolism like HIF1α and the protein kinase B (AKT) signaling pathways, which are involved in upregulating glycolysis [66]. Further, MS patients have significant changes in serum levels of different metabolites, including an increase in serum nicotinamide adenine dinucleotide (NADH) compared to healthy patients. Interestingly, increased concentrations of glutamine and glutamate has been found in the cerebrospinal fluid and brain biopsies from affected individuals. Increases in these metabolites were also associated with disease severity and have been suggested to be potential biomarkers for MS [64–66]. In MS lesions, mitochondrial changes have also been reported, including a loss of complex I and IV activity in the mitochondrial ETC, implicating a role for mitochondrial damage and oxidative stress in MS disease pathogenesis [67–72]. While a number of metabolic differences exist between MS patients and healthy controls based on what has been reported in the current literature, these studies have not focused on the metabolic characteristics of CD4+ T, which similar to T1D and lupus, are the main effectors in driving autoimmunity in MS. In 2015, De Riccardis et al. set out to determine the metabolic phenotype of immune cells in the MS autoimmune response using CD4+ T cells isolated from peripheral blood mononuclear cells (PBMCs) of relapsing remitting MS patients [68]. They found that CD4+ T cells from these patients had impaired OXPHOS activity and decreased activity of mitochondrial complex 1 and IV compared to healthy subjects, confirming the findings from earlier studies [68,69]. When analyzing glucose metabolism in CD4+ T cells from these patients, MS CD4+ T cells displayed increased glycolytic flux as measured by increased activities of two major rate limiting enzymes in the glycolysis pathway, hexokinase and phosphofructokinase [68]. During activation, CD4+ T cells switch to a program dominated by aerobic glycolysis, which is the conversion of glucose to lactate despite the presence or availability of oxygen. Because of this phenomenon, lactate secretion is a useful output to measure reliance on glycolytic metabolism in T cells. As expected, extracellular lactate release was enhanced in T cells from MS patients compared to control subjects [68]. This, along with the findings that CD4+ T cells from MS patients had reduced ratios of ATP from mitochondrial metabolism over glycolytic ATP, as well as increased glucose transporter 1 (Glut1) expression (the most important glucose transporter for T cells) compared to healthy controls indicates an increased reliance on glycolysis by autoreactive T cells in MS [68]. Interestingly, the enhanced reliance on glycolysis by circulating CD4+ T cells from MS patients supplies further evidence that autoreactive T cells have enhanced bioenergetics compared to T cells from healthy controls. However, there are limitations to this study, which included total CD4+ T cells. It would be interesting to note whether the enhanced glycolytic metabolic phenotype described was due to an increase in Teffectors in the periphery in those with MS compared to healthy individuals, or whether metabolism was truly dysregulated in naïve, effector, and memory T cell populations between MS patients and healthy subjects, as performed in work described earlier in lupus [53,68].
Contrary to the findings by De Riccardis et al., another study found that T cells from MS patients were in fact less glycolytic and had impaired engagement of mitochondrial respiration than their healthy counterparts, as indicated by a lower extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) measured by Seahorse Extracellular Flux analysis [73]. These measurements are indicators of glycolysis and OXPHOS, respectively. This metabolic defect was confirmed by measuring the expression of key enzymes involved in both glycolysis and mitochondrial respiration. Reduced protein levels of key enzymes such as Glut1 and hexokinase, correlated with the seahorse analysis performed, further confirming a metabolic defect in CD4+ T cells from MS patients [73]. This finding is in conflict with the enhanced bioenergetic profile of T cells observed in MS patients from the study outlined above, and could be due to differences in the patient cohort, as the T cells in the De Riccardis study were from patients that had been treated in the past, albeit these patients had not been treated for at least 3 months [68]. On the contrary, samples from the patients in the La Rocca study were isolated from affected individuals who had not underwent treatment for their condition. This difference in treatment regimens between the two studies could account for these metabolic differences, as immunomodulatory agents have been shown to modulate cellular metabolism [73]. Moreover, the methods used to assess metabolism in both studies were vastly different, as the De Riccardis study relied on the evaluation of enzymatic activity and the La Rocca study utilized an approach that took the whole cell into consideration using a functional analysis like Seahorse Extracellular flux. Regardless, the impaired metabolic profile of MS CD4+ T cells in the La Rocca study is an interesting finding, as glycolysis is required not only for T cell differentiation but also for acquisition of specialized effector functions, like IFNγ secretion [74]. Further, this is different than the metabolic phenotype of autoreactive T cells from other autoimmune diseases in that instead of having enhanced bioenergetics, T cells from these patients had reduced glycolysis and OXPHOS. How these T cells are still able to become activated and perpetuate disease under a reduced bioenergetic state warrants investigation so as to obtain a full picture of the metabolic choices T cells have at their disposal. Further, the activation status of these T cells would also be important to investigate. It has been reported in the cancer literature and other autoimmune disorders that a chronic exposure to antigen and metabolic competition in the tumor microenvironment (TME) can lead to T cell exhaustion or anergy [75]. T cell exhaustion in particular is associated with increased expression of checkpoint molecules like programmed cell death protein 1 (PD-1) and lymphocyte activation gene-3 (Lag-3), which are capable of reprograming T cell metabolism themselves, and will be discussed in more detail below [76,77]. Further characterization of the expression of key surface proteins and delineation of the molecules that regulate metabolism and cell fate decisions should be further explored, in addition to understanding the activation status. Finally, it is vital to our understanding of autoimmune disease pathogenesis to study metabolic profiles of T cells over the course of autoimmune destruction. These studies are important to better understand the metabolic dependencies of autoreactive T cells over the course of disease pathogenesis and will aid in the discovery of novel biomarkers and pathways that can be solicited to specifically target these pathogenic T cells therapeutically to prevent or reverse disease in affected individuals.
Rheumatoid ArthritisIn RA, altered immune tolerance and metabolic defects contribute to chronic synovial inflammation and joint destruction, mediated in large part by CD4+ T cells [78,79]. The first evidence that altered bioenergetics played a role in RA pathogenesis was found in early studies analyzing serum samples from RA patients, where aldolase A and α-enolase, two enzymes of the glycolytic pathway, were identified as autoantigens [15,80,81]. Similarly to the results described above, CD4+ T cells from a spontaneous mouse model of RA were more metabolically active compared to controls. Further, glycolysis inhibition with 2-DG significantly reduced joint inflammation and activation of adaptive and innate immune cells, as well as autoantibody production [82].
Contrary to the findings in animal studies, naïve CD4+ T cells isolated from the PBMCs of RA patients were found to be energy deprived and were unable to fully engage aerobic glycolysis [78]. This was indicated by reduced glucose utilization, lactate production, and generation of intracellular ATP 72 hours post in vitro stimulation, and correlated with increased apoptosis, which was 50% higher in RA T cells compared to controls [78]. To understand whether RA T cells were anergic thus leading to reduced glucose utilization, parameters of T cell responsiveness were monitored post stimulation. Interestingly, RA T cells responded as vigorously as their control counterparts, proliferated faster, and were able to produce IL-2 to the same extent as control T cells, indicating that naïve RA T cells were able to respond to environmental stimuli and undergo activation at a rate comparable or better than control T cells, however metabolically speaking RA T cells displayed abnormalities in the utilization of glucose, ATP production, and were more susceptible to undergo cell death in response to stimulation [78]. This metabolic phenotype was found to be due to a defect in up regulating the rate limiting enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), a key regulator of glycolytic flux [78].
The immune microenvironment plays a significant role in T cell differentiation and function, as evidenced by studies researching the impact the TME has on tumor-infiltrating lymphocyte (TIL) function. In order to determine whether the failure to induce PFKFB3 expression in RA T cells was a consequence of the inflammation associated with RA disease progression, PFKFB3 transcript levels in CD4+ T cells were correlated with a disease activity score, indicating disease severity. Contrary to what you would expect, patients with lower disease activity had no advantage over patients with more severe disease [78]. Further, a failure to induce PFKFB3 expression did correlate with diagnosis of RA, but not with the inflammatory milieu, indicating that reduced PFKFB3 expression by RA T cells is a defect in all RA patients regardless of disease clinical score or severity [78]. Nonetheless, it’s important to note that autoimmunity precedes the onset of clinical symptoms, and that exposure of CD4+ T cells to chronic stimulation in an inflammatory environment likely plays a key role in altering immune function and lymphocyte stability in the local micromilieu. For example, in the TME, a competition for nutrients, especially glucose between tumor cells and tumor infiltrating lymphocytes, is thought to drive T cell dysfunction and exhaustion thus leading to cancer persistence and tumorigenesis [75]. This too could be the case in RA, as the fibroblast-like synoviocytes (FLS) that play a major role in the initiation and development of synovial inflammation and joint destruction in patients display an activated phenotype characterized by enhanced glycolytic metabolism similarly to cancer cells [79,83]. This enhanced metabolic profile allows RA FLSs to exhibit an abnormal capacity for migration, invasion, and proinflammatory cytokine production that contribute to disease pathology [79]. The proinflammatory nature of FLSs in RA most likely promotes a hostile immunosuppressive environment that over time, renders autoreactive infiltrating T cells dysfunctional due to high lactate output and a lack of nutrients available to fuel proper T cell activation [47,75]. Therefore, its plausible that the metabolic defects in CD4+ T cells from RA patients does not cause disease, rather it occurs as a consequence of the environment. Indeed, treatment of RA FLSs with the anti-glycolytic PFK15, a competitive inhibitor of the enzyme PFKFB3, lead to decreased expression of proinflammatory cytokines, migration, and proliferation [79]. Whether metabolic regulation of RA FLS lead to normalization of T cell metabolism and function remains unknown and should be investigated more thoroughly. Moreover, more studies are needed to fully dissect the metabolic features of CD4+ T cells in RA from disease onset through progression. This would allow for thorough examination of the metabolic defects plaguing autoreactive T cells in RA, and may provide new details that explain differences seen in mouse models and patient samples.
While metabolism as a therapeutic target has been exploited in many autoimmune diseases, such as the ones detailed in this review, it has been largely unexplored in T1D research. However, like CD4+ T cells in lupus, MS, and RA, there is growing evidence from our laboratory and others that β cell reactive T cells demonstrate enhanced bioenergetic demands and metabolic defects that drive autoimmune targeting of the pancreatic β cell. While most of this review has focused on utilization of glycolysis to fuel T cell activation, mitochondrial OXPHOS and ROS are also enhanced during activation, as mitochondrial derived ROS not only act as a third signal for efficient T cell activation , but also are essential for IL-2 production and proliferation upon antigenic stimulation [84,85]. T cell mitochondrial dysfunction is seen often in autoimmunity, and is attributed to increased mitochondrial inner-membrane hyperpolarization (MHP) [84]. Recently it has been identified that the production of ROS and synthesis of ATP are tightly regulated by maintenance of mitochondrial membrane potential. Further, mitochondrial dynamics and function are thought to drive T cell differentiation [86,87]. More specifically, it has been demonstrated that increased mitochondrial membrane potential leads to an effector T cell phenotype characterized by increased glycolysis and ROS production [88]. Interestingly, T cells from patients with T1D display MHP and increased mitochondrial membrane potential, indicating a more activated phenotype compared to healthy control T cells [84]. This phenotype was seen in T cells in the CD4 and CD8 compartment regardless of activation status, indicating that MHP is a general characteristic of T cells in patients with T1D. Moreover, enhanced MHP was linked to increased ROS production and IFNγ secretion, indicating that mitochondrial dysfunction promotes effector functions of autoreactive T cells due to an altered proinflammatory T cell effector response [84]. This finding that ROS drive T cell activation and effector capabilities correlate with data from our laboratory, where use of a manganaese metalloporphyrin (MnP) with ROS scavenging capabilities during T cell activation inhibited diabetogenic CD4+ T cell responses in vitro and delayed diabetes progression in an adoptive transfer model of T1D in vivo [18,20,89–91]. Work by Previte et al. demonstrated that redox modulation via MnP treatment during activation maintained AMP-activated protein kinase (AMPK) activation that resulted in decreased mTOR activation and a reduced ability to undergo metabolic reprogramming to aerobic glycolysis [91]. These data demonstrated that ROS are required for driving and sustaining T cell activation and promoting the metabolic switch to glycolysis [91]. Altogether, this data suggests that self-reactive CD4+ T cells have enhanced bioenergetic demands fueled by glycolysis, and that this increased reliance on the glycolytic pathway long-term induces MHP, increases ROS production, and makes these T cells super-effectors that drive Th1 mediated β cell destruction. Moreover, the fact that T1D T cells secrete more IFNγ after activation is in itself indicative of an enhanced glycolytic profile, as aerobic glycolysis controls preferential translation of IFNγ mRNA and is required for acquisition of T cell effector functions [74]. Finally, data presented by Chen et al. indicating that naïve T cells had higher MHP supports the idea that autoreactive T cells are in a heightened activation state. This data, along with the potential for poor peptide binding due to dysfunctional antigen presentation, may provide further insight as to how and why autoreactive T cells are able to escape deletion in the thymus and go unabated in the periphery [84,92]. It may also be a reason as to why effector T cells become resistant to Treg mediated control or explain why Treg function is defective in T1D, as enhanced glycolysis may lead to FoxP3 destabilization and subsequent loss of suppressive capabilities.
Hyperglycemia and the Immune Microenvironments Role in Shaping β Cell Reactive T Cell Responses in T1DA common theme discussed thus far has been understanding the role of the immune microenvironment on T cell differentiation and function, and how that contributes to autoimmune disease progression and pathology. As glucose is the substrate of choice for T cells undergoing activation and is required for metabolic reprogramming to occur, it would be interesting to know whether hyperglycemia contributes to the heightened bioenergetic profile and metabolic defects of β cell reactive CD4+ T cells in T1D. One could postulate that autoreactive T cells in T1D, which appear to be more glycolytic compared to T cells from healthy controls, are able to utilize glucose more freely due to its abundant availability in the blood, thereby enforcing a proinflammatory program that leads to a predominate effector T cell phenotype. This phenomenon has been previously described for CD8+ T cells, where in vitro simulated hyperglycemia increased IFNγ secretion [93,94]. In human studies, Chen et al analyzed the impact of hyperglycemia on T cell mitochondrial membrane potential by measuring membrane potential in T cells from a cohort of patients with Type 2 Diabetes (T2D) and comparing it to the membrane potential of T cells from patients with T1D [84]. This comparison allowed for delineating whether the high glucose environment or immune abnormalities associated with autoimmune conditions lead to the metabolic defects in T cells from T1D patients. Intriguingly, it was found that T cells from T2D had membrane potential levels that were indistinguishable from controls. However, this was significantly lower than the membrane potential of T cells isolated from T1D patients. These data indicate that the mitochondrial defects seen in T cells from individuals affected by T1D is a consequence of autoimmunity, and not a consequence of the high blood glucose levels and loss of glucose control in these patients [84]. Whether T cells between T1D and T2D were more glycolytic in hyperglycemic environments remains unknown. It would be intriguing to follow metabolic and mitochondrial dysfunction in autoreactive T cells starting during development in the thymus through early initiation of autoimmunity, all the way to onset and progression of clinical symptoms and disease. While this would be difficult to do using patient samples, a large-scale study following the course of disease could be more easily done using the NOD mouse, a spontaneous model of T1D that closely mimics key characteristics of human disease. While not a perfect system, it would allow for a better understanding of how T cells respond to various environmental stimuli encountered during development and in the periphery that could contribute to metabolic and immunologic dysfunctions.
Furthermore, defective Treg function is a common feature of autoimmune diseases [22,36,95]. Metabolism plays a key role in regulating the balance between Teffectors and Tregs [5,34,37–39,42,46]. While Treg suppressive capabilities and FoxP3 stability are thought to be supported by use of FAO, glycolysis has been demonstrated to be required for Treg proliferation and circulation [39,49]. Glycolysis, which is associated with Teffector activation, drives proinflammatory responses while OXPHOS and FAO are associated with Tregs, and drive an anti-inflammatory response. Modulating the glycolysis pathway has been shown to inhibit effector T cell responses while also promoting the induction of FoxP3+ Tregs [10,39,46,47,51–53,82]. In SLE, Treg cells exhibit increased mTOR activity that correlated to their dysfunction and diminished suppressive capacities, which could be corrected with rapamycin treatment [61]. This provides evidence that Tregs in autoimmune conditions are more glycolytic, and could indicate why they fail to control autoreactive T cells in SLE, MS, RA, and T1D [61]. Notably, while rapamycin treatment is capable of expanding SLE Tregs and correcting functional defects, mixed results have been obtained with Treg expansion in T1D patients [55–57,59–61,96]. One study by Monti et al. investigating the effect of rapamycin on human Tregs in vivo demonstrated that treatment did not alter the frequency, proliferation, or cytokine production of circulating Tregs, but it enhanced their capability to suppress proliferation of effector T cells in T1D [96]. The varying effects of rapamycin on Treg expansion seen in SLE and T1D could be attributed to the type of pathogenic T cells that mediate autoimmune damage in both conditions. In SLE, patients have enhanced Th17 cell responses that correlate with disease activity [97]. On the other hand, T1D is a primarily Th1 mediated disease [18,20,21,23,89]. This difference in Th cell differentiation could indicate why Treg expansion is more easily accomplished in SLE patients. In particular, recent literature has indicated that T cell metabolism, namely the glycolysis pathway, is an important checkpoint controlling the differentiation of Th17 and Treg cells, as blocking glycolysis has the ability to inhibit Th17 differentiation while promoting Treg cell generation [39,46]. Further research is needed to dissect the metabolic features of effector T cells and Tregs in autoimmune conditions, with particular attention to how T cells in various disease pathologies differ from one another. Specifically in T1D, It would be of interest to know whether high blood glucose levels and enhanced glycolysis leads to destabilization of the Treg lineage, and to further characterize whether Treg dysfunction in autoimmunity is a result of metabolic alterations, as appears to be the case in SLE [61].
Enforcing T Cell Exhaustion: A Foe in Cancer, a Friend in Autoimmunity?Immune dysfunction in cancer is thought to occur as a result of T cell exhaustion and increased Tregs in the tumor microenvironment that limit anti-tumor immunity. T cell exhaustion, which is typically associated with CD8+ T cells but can occur in the CD4 compartment, is characterized by loss of effector functions, altered cytokine responses, overexpression of checkpoint inhibitory receptors (i.e., PD-1, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), Lag-3, and T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), and altered metabolic programs leading to T cell hyporesponsiveness and cancer progression [98]. Checkpoint blockade of inhibitory receptors, especially PD-1 have revolutionized cancer immunotherapy, as they have proven to be effective in reinvigorating exhausted T cells in the TME. Unfortunately, immunotherapy is known for a multitude of immune-related adverse effects, including onset of autoimmune disorders like PD-1 inhibitor induced T1D [99]. Specific work in T1D has revealed that PD-1/Programmed death-ligand 1 (PD-L1) deficiency and blockade or deletion of Lag-3 in NOD animals accelerates the onset of autoimmune diabetes, indicating an important role for checkpoint inhibitory receptors in regulating T cell responses in T1D [100–102].
Studies using mouse models and samples from human patients have revealed that the PD-1/PD-L1 pathway is dysregulated in T1D [103–105]. Indeed, activated T cells from new-onset T1D patients fail to upregulate PD-1 compared to healthy subjects [104]. This was true too for CD4+ T cells in adults with long-standing T1D [106]. Interestingly, this phenotype was seen only in CD4+ T cells from T1D patients, whereas PD-1 expression was similar between T cells from SLE patients and healthy controls , indicating PD-1 dysregulation is specific to T1D onset and pathogenesis [106].
Lag-3 is another checkpoint molecule that has been tied to immune cell dysfunction in T1D. This was confirmed in work by Bettini et al. where deletion of Lag-3 in autoimmune prone NOD animals resulted in accelerated diabetes with 100% incidence that corresponded to increased T cell islet infiltration, and not due to Treg dysfunction [101,107].This phenotype was exclusive to autoimmunity, as Lag-3 deletion in non-autoimmune prone C57BL/6 mice resulted in a minor phenotype with no development of spontaneous disease, similarly to the minimal immunopathologies associated with PD-1 knockout in non-autoimmune prone mice [108,109]. Together, these data indicate a critical role for Lag-3 in controlling early expansion of autoreactive T cell clones. It would be important to study Lag-3 expression on T cells from healthy individuals and T1D patients to better understand whether Lag-3, like PD-1, is dysregulated during autoimmunity. Due to the synergistic nature of PD-1 and Lag-3, we would hypothesize that Lag-3 expression is decreased or signaling is dysfunctional in T cells from T1D patients. Moreover, it’s clear that checkpoint inhibitory receptors, including both PD-1 and Lag-3, are important for maintaining self-tolerance and may be critical mediators that drive autoimmunity.
As one would expect, the state of T cell exhaustion is accompanied by distinct metabolic features [110]. Moreover, both PD-1 and Lag-3 have been implicated in reprogramming lymphocyte metabolism. For example, PD-1 signaling specifically promotes lipolysis and FAO and leaves T cells unable to engage in aerobic glycolysis via stabilization of phosphatase and tensin homolog (PTEN) [76]. As glycolysis is required for acquisition of T cell effector functions, this finding makes sense as exhausted T cells expressing PD-1 are dysfunctional in their ability to secrete proinflammatory cytokines and have limited effector capabilities [98]. This reliance on FAO is associated with T cell longevity and enhanced survival while exhausted and unable to utilize other key nutrients [76,111]. A paper published this past year by our laboratory investigated the role of Lag-3 in maintaining metabolic quiescence in naïve CD4+ T cells [77]. Interestingly, it was determined that Lag3 -/- CD4+ T cells demonstrated greater glycolytic capacity and enhanced proliferation and effector functions (i.e., IFNγ secretion) during activation, indicating a role for Lag-3 in regulating T cell activation and clonal expansion. This is an interesting finding and may provide evidence for enhanced metabolic phenotypes of T cells in autoimmune conditions, as inhibitory receptor expression is impaired in diabetogenic autoreactive T cells [104]. Impaired expression of PD-1 and Lag-3 could explain the hyperactivated phenotype and enhanced effector capabilities of autoreactive CD4+ T cells that are responsible for disease-inducing potential of these cells. Finally, altered bioenergetics and enhanced glycolysis in diabetogenic CD4+ T cells may confer resistance to Treg mediated suppression or lead to Treg dysfunction that is known to occur during T1D pathogenesis [21,22,26,34,36]. Based on these data, it would be interesting to know whether recapitulating an environment like the TME in cases of autoimmunity could prevent or reverse disease. As T cells in autoimmune prone individuals fail to upregulate these inhibitory receptors, enforcing the checkpoints and modulating T cell metabolism to promote T cell exhaustion may lead to tolerance in affected individuals. In fact, a study by Tilstra et al. demonstrated that kidney-infiltrating T cells in a mouse model of lupus nephritis become metabolically and functionally exhausted after organ infiltration as a means to limit damage to self [112]. Therefore, enforcing this phenotype prior to disease onset via metabolic regulation may provide a new way to limit autoimmune disease associated pathologies [112]. Further investigation is needed to determine whether glycolysis inhibition or promotion of FAO leads to an exhausted state.
Current therapeutic strategies for T1D have focused on 2 different approaches including (1) restoring or preserving endogenous β cell mass or (2) combatting autoreactivity and autoimmunity. While efforts to restore and preserve β cell mass have been interesting and shown promise in preclinical studies, these strategies ultimately fail due to a recurrence of autoimmunity [113,114]. The first proof of principle study showing that immunotherapy is effective in human patients with T1D was published in 1984, and investigated the use of Cyclosporine in recent onset T1D patients [115,116]. This has given precedence for finding ways to limit the autoimmune destruction of β cells by the immune system. Two clinical trials have been published this year that investigated the use of low dose anti-thymocyte globulin (ATG) in new onset T1D patients and the anti-CD3 antibody Teplizumab in relatives at risk for developing disease and found that both low-dose ATG preserved β cell mass while Teplizumab delayed disease onset [117,118]. While disease was successfully delayed and β cell mass was preserved for a period of time, both treatments consequently lead to lymphopenia in treated patients [117,118]. Moreover, these new studies, while effective, have not addressed the broad immunosuppression associated with the treatment. This is a major hurdle to overcome especially in T1D, which affects juvenile patients, as wiping out the immune system could have broad implications for the ability to overcome potential infections and pathogens. Instead, they have used newer therapeutics that have the same lymphopenic affect that was demonstrated 35 years ago with Cyclosporine. Importantly though, these studies, along with the ability to predict onset of T1D based on the presence of one or more autoantibodies, has given precedence for manipulating T cell responses to prevent or delay disease onset in those predisposed to autoimmunity.
As discussed in detail in this review, modulating CD4+ T cell metabolism has the ability to both prevent and reverse autoimmune conditions [46,51–54,82]. While not yet explored in T1D, the potential of metabolic intervention to be successful to control islet-antigen specific T cell responses exists and deserves further examination. At the time of diagnoses, patients with T1D have already experienced longstanding autoimmune onslaught to pancreatic β cells, and thus only have approximately 10–20% β cell mass still present [1,3,4,21]. As in most autoimmune conditions, autoimmunity (specifically activation of autoreactive CD4+ T cells) precedes downstream damage, and is an important consideration to make when thinking about where and when metabolic intervention is best suited. Studies in T1D have shown that immunotherapy is successful in delaying and/ or preventing onset [115–118]. Based on the immunopathology of T1D, and due to the importance of CD4+ T cells in disease initiation and progression, we would hypothesize that use of metabolic inhibitors would be best suited as a prophylactic strategy, prior to diagnoses. Genetic predisposition and the presence of multiple autoantibodies can predict who will progress to have T1D [1,4,21,117,118]. Although not currently used in the clinic, testing for the presence of autoantibodies to β cell antigens in children with a family history of T1D or in those at risk to developing autoimmunity could be used to determine whether metabolic intervention should be initiated. Moreover, the natural history of T1D takes place over a number of years, giving ample time for intervention prior to complete loss of dynamic glucose sensing and insulin secretion due to β cell loss [1]. This idea of disease prevention was outlined above. Studies in SLE have demonstrated the success of metabolic modulators in preventing autoimmunity [52–54]. Also, while it is possible that chronic administration may be necessary, inhibiting glycolysis has been shown to promote Treg differentiation while inhibiting effector T cell expansion [47,52–58,61–62]. If metabolic intervention can restore the balance between effector T cells and Tregs, and improve suppressive function of dysfunctional Tregs in autoimmune diseases, a shorter treatment regimen may be successful in establishing durable tolerance to self-antigens. However, more work is needed to fully understand the ability of metabolic inhibitors to prevent or reverse disease, especially T1D, in order to determine length of treatment needed to establish tolerance.
On the contrary to immune-targeted therapies that result in repression of global immunity, metabolic therapies have demonstrated great success in selective targeting of self-reactive T cells while leaving humoral immunity unscathed and intact [12,51–54,82]. This specific targeting of autoreactive T cells via use of anti-metabolites even when administered systemically is a phenomenon termed “cellular selectivity based on demand”, and was first described by Lee et al, in a study where targeting glucose and glutamine metabolism specifically prevented allograft rejection [119–121]. The idea behind this theory is that use of generic inhibitors of universal metabolic processes selectively affect cells with the greatest metabolic demand, without altering normal cellular homeostatic function [119–121]. While effector T cells alone have increased metabolic demands to support activation, proliferation, and effector functions, research in autoimmunity has provided evidence that autoreactive T cells have altered bioenergetic programs that allow for their increased effector capabilities. These metabolic abnormalities could contribute to their persistence in the periphery and disease-promoting capabilities compared to T cells from healthy individuals. This makes metabolism an ideal target for autoimmunity, where enhanced metabolically demanding programs that self-reactive T cells rely on can be specifically modulated to promote tolerance. Moreover, as immune cells utilize distinct metabolic programs, inhibition of glycolysis for example should in theory have minimal effects on Tregs or established memory T cells to vaccines. When using metabolic inhibitors, like 2-DG, to alleviate autoimmune targeting by CD4+ T cells as described in detail above, the remainder of the immune system is otherwise unharmed and functionally able to respond appropriately to non- self-antigens (i.e., to immunization). However, it’s important to consider that metabolic intervention may also modulate immune cells other than CD4+ T cells. All immune cells, including macrophages, dendritic cells, B cells, and CD8+ T cells, undergo metabolic reprogramming to aerobic glycolysis upon activation [9,10,122,123]. Therefore, its plausible that use of inhibitors like 2-DG, metformin, or PFK15 could also alter APC, B cell, or CD8+ T cell function. In fact, treatment with glycolysis inhibitors like 2-DG in mouse models of SLE and T1D resulted in reduced CD8+ T cell expansion and effector function and also lead to reduced autoantibody production by B cells [51,53,124]. All in all, current immunotherapeutic strategies to combat T1D have had harsh repercussions on the entire immune system. Moreover, there is evidence that autoreactive T cells do not play by the same rules metabolically and have altered metabolic demands and requirements compared to their non-autoimmune counterparts. Whether abnormal metabolism is a feature of other immune cell compartments in autoimmunity warrants further investigation. Moreover, further work is needed to determine the impact of metabolic modulators on CD8+ T cell, B cell, and APC function, as most of the studies done thus far have focused on modulating CD4+ T cell metabolism. The importance of CD4+ T cells in autoimmunity and their distinct metabolic features provide a unique opportunity to move immunotherapy towards targeting metabolism, which has a proven track record of specifically controlling rogue self-reactive T cell responses without interfering with basal cellular metabolic programs of other cells in the body or serious side effects.
T1D is an autoimmune disease that ultimately results in hyperglycemia due to CD4+ T cell driven attack of the pancreatic β cell. Recent work in the field of immunometabolism has demonstrated that T cell function is regulated by bioenergetics and cellular metabolism. Consequently, metabolic programs have the ability to influence the final outcome of an immune response, specifically in terms of whether a pro- or anti- inflammatory response will ensue This is especially true in autoimmune settings, where dysregulated T cell metabolic programs lead to aberrant T cell activation, proliferation, resistance to Treg mediated suppression, and enhanced proinflammatory cytokine production and effector capabilities. While immunomodulatory strategies for prevention and treatment of T1D have shown promise in the clinic, they lack specificity in their ability to target the cells of interest, leading to low T cell counts and compromised immunity to pathogens and foreign threats. This is especially dangerous in T1D, where children are the primary patients affected. Due to the importance of T cell metabolism in dictating immunity, anti-metabolites and inhibitors of the major metabolic pathways that drive T cell responses are being used to combat autoimmunity and restore tolerance in affected individuals (Table 1). While still largely unexplored in T1D, modulating T cell metabolism in other autoimmune diseases like lupus, RA, and MS have successfully prevented and reversed disease in animal models, and in some instances has led to increased Treg function and control of Teffectors, which ultimately fail to maintain peripheral tolerance in afflicted individuals. These data indicate that metabolic regulation has the ability to restore the balance between glycolysis and OXPHOS that ultimately has the ability to induce durable tolerance in those predisposed to autoimmunity (Figure 4).
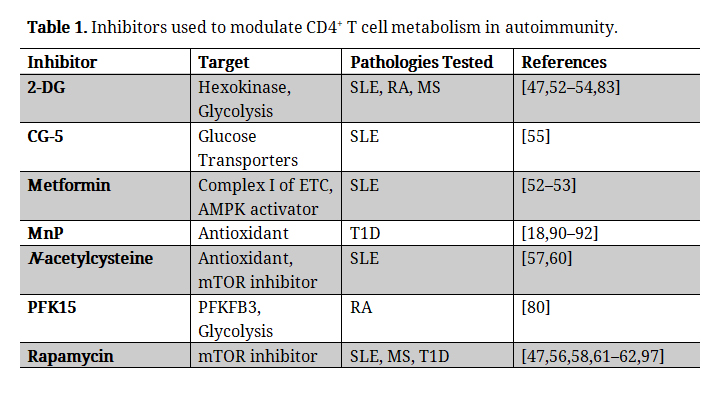 Table 1. Inhibitors used to modulate CD4+ T cell metabolism in autoimmunity.
Table 1. Inhibitors used to modulate CD4+ T cell metabolism in autoimmunity.
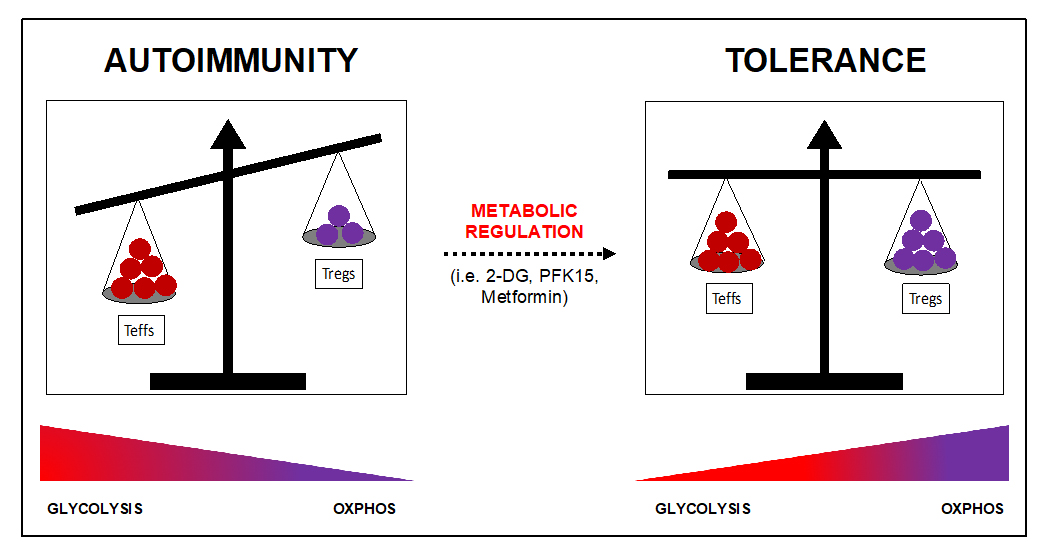 Figure 4. Metabolic regulation promotes tolerance by restoring the balance between effector T cells and Tregs. Escape of autoreactive T cells from deletion during thymic development, activation of autoreactive T cells, and defective Treg controls of Teffectors in the periphery results in autoimmunity. While glycolysis is associated with a pro-inflammatory response, OXPHOS controls anti-inflammatory responses associated with tolerance and regulation. Metabolic regulation and use of specific inhibitors of ubiquitous metabolic pathways, such as 2-DG, can inhibit Teffector responses and promote Treg stability and function that promote tolerance and prevent or reverse autoimmune associated pathologies.
Figure 4. Metabolic regulation promotes tolerance by restoring the balance between effector T cells and Tregs. Escape of autoreactive T cells from deletion during thymic development, activation of autoreactive T cells, and defective Treg controls of Teffectors in the periphery results in autoimmunity. While glycolysis is associated with a pro-inflammatory response, OXPHOS controls anti-inflammatory responses associated with tolerance and regulation. Metabolic regulation and use of specific inhibitors of ubiquitous metabolic pathways, such as 2-DG, can inhibit Teffector responses and promote Treg stability and function that promote tolerance and prevent or reverse autoimmune associated pathologies.
Moreover, it has been determined that at the resolution of disease pathologies in autoimmune diseases, T cells are metabolically and functionally exhausted, similarly to that observed in the tumor microenvironment. Interestingly, the classical immune checkpoints, such as PD-1 and Lag-3, have been implicated in controlling T cell expansion and function, but are less able to be expressed by T cells from T1D patients compared to healthy controls. Therefore, targeting metabolism to force T cells into a state of exhaustion may be a novel therapeutic strategy that is able to control autoreactive T cell activation and downstream autoimmune damage. One concern associated with metabolic modulation is that these therapeutics will also affect basal metabolism in all cells of the body. Interestingly, this does not seem to be the case, as systemic administration of these inhibitors has shown selective targeting capabilities with no adverse effects on global immunity or other cells. This phenomenon has been deemed cellular selectivity based on demand, whereas only the most metabolically active cells are affected by administration of ubiquitous anti-metabolic drugs like the glycolysis inhibitor 2-DG. Whether this holds true in human patients remains unknown and needs to be explored, however the data from mouse studies are compelling and demonstrate a selective targeting that is unprecedented when administering a drug systemically. However, more research, especially in T1D is needed to fully understand the metabolic defects that lead to escape and persistence of self-reactive T cell clones that result in the autoimmune pathologies described within this review. It is also vital to determine how durable of a tolerance is achieved by metabolic regulation, as recurrence of autoimmunity remains a threat and concern.
Finally, although this review specifically discussed targeting CD4+ T cell metabolism, a number of immune cells including macrophages, dendritic cells, B cells, and CD8+ T cells play critical roles in driving attack of the pancreatic β cell; all of which undergo metabolic changes during their subsequent activation. Further work is necessary to understand whether metabolic pathways are dysregulated in APCs and CD8+ T cells during autoimmunity, and whether these cells too are affected by metabolic intervention. All in all, it appears targeting metabolism has the potential to be the next wave in immunotherapies used to control islet-reactive CD4+ T cells.
The authors declare that they have no conflicts of interest.
This research was funded by the Juvenile Diabetes Research Foundation (JDRF), grant number 2-SRA2020-910-S-B awarded to JDP, a Cochrane Weber grant from UPMC Children’s Hospital of Pittsburgh Foundation awarded to JDP, and a Research Advisory Committee Predoctoral Fellowship from UPMC Children’s Hospital of Pittsburgh awarded to CPM.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
Martins CP, Piganelli JD. Targeting T cell Metabolism to Combat Autoimmunity: Implications for the Future of Type 1 Diabetes Therapeutics. Immunometabolism. 2020;2(2):e200010. https://doi.org/10.20900/immunometab20200010

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




