Location: Home >> Detail
J Psychiatry Brain Sci. 2020;5:e200002. https://doi.org/10.20900/jpbs.20200002
 ,
Sylvain Bouix 2,3,
Dost Ongur 1,2,
Martha E. Shenton 2,3
,
Sylvain Bouix 2,3,
Dost Ongur 1,2,
Martha E. Shenton 2,3
1 Schizophrenia and Bipolar Disorder Program, McLean Hospital, Belmont, MA 02478, USA
2 Department of Psychiatry, Harvard Medical School, Boston, MA 02215, USA
3 Psychiatry Neuroimaging Laboratory, Brigham and Women’s Hospital, Boston, MA, 02215, USA
† This work was supported by grants from the National Institute of Mental Health to KEL (R01MH117012) and MES (U01MH109977)
* Correspondence: Kathryn E. Lewandowski.
Psychotic disorders are severe, debilitating, and even fatal. The development of targeted and effective interventions for psychosis depends upon on clear understanding of the timing and nature of disease progression to target processes amenable to intervention. Strong evidence suggests early and ongoing neuroprogressive changes, but timing and inflection points remain unclear and likely differ across cognitive, clinical, and brain measures. Additionally, granular evidence across modalities is particularly sparse in the “bridging years” between first episode and established illness—years that may be especially critical for improving outcomes and during which interventions may be maximally effective. Our objective is the systematic, multimodal characterization of neuroprogression through the early course of illness in a cross-diagnostic sample of patients with psychosis. We aim to (1) interrogate neurocognition, structural brain measures, and network connectivity at multiple assessments over the first eight years of illness to map neuroprogressive trajectories, and (2) examine trajectories as predictors of clinical and functional outcomes. We will recruit 192 patients with psychosis and 36 healthy controls. Assessments will occur at baseline and 8- and 16-month follow ups using clinical, cognitive, and imaging measures. We will employ an accelerated longitudinal design (ALD), which permits ascertainment of data across a longer timeframe and at more frequent intervals than would be possible in a single cohort longitudinal study. Results from this study are expected to hasten identification of actionable treatment targets that are closely associated with clinical outcomes, and identify subgroups who share common neuroprogressive trajectories toward the development of individualized treatments.
Psychotic disorders, including schizophrenia (SZ), schizoaffective disorder (SZA), and bipolar disorder with psychosis (BDP) are severe, debilitating, and even fatal [1] and are a leading cause of disability world-wide [2]. Unfortunately, a majority of patients with psychosis experience functional impairments, even after symptom remission [3,4], underscoring further the need for effective, targeted treatments to improve outcomes. The development and implementation of targeted and effective treatments for psychosis is critically dependent on a clear understanding of the timing and nature of disease progression in order to target processes amenable to intervention.
Evidence of early and ongoing disease-related changes in brain and neurocognitive measures in psychosis, often referred to as “neuroprogression,” including cognitive dysfunction, gray matter reduction and ventricular enlargement, and regional structural and connectivity alterations, have been described using multiple modalities including neurocognitive testing, PET, CT, and fMRI imaging techniques, and post-mortem brain studies (Table 1) [5–93]. However, these studies rely mainly on cross-sectional data comparing groups at particular illness stages (e.g., high risk, first episode, established) to healthy controls or to each other. Existing longitudinal studies typically involve only two measurement points, or repeated assessment years apart, thereby limiting our understanding of the actual trajectories and key inflection points of these disease markers [94]. Thus, while it is evident that significant progressive brain changes occur during the years following an initial episode, the timing and course of progression across domains remains unclear. This knowledge gap limits our ability to develop interventions that capitalize on plasticity in key systems during this critical and dynamic period of illness, and hinders the development of targeted treatments when they may be most effective, potentially preventing further decline and chronic loss of functioning.
Early Psychosis: A Critical PeriodThe years after illness onset represent a critical period where early intervention strategies may be most effective, before irreversible brain alterations take place. Effective treatment after a first episode of psychosis (FEP) not only improves functioning but may actually alter illness trajectories placing patients on a path toward recovery [95]. Disease trajectories appear to crystalize in the years following an initial episode of illness, making this a critical period for intervention after which effectiveness may be greatly reduced [96,97]. While much is known about neuroprogressive changes in FEP and established illness, less is known about the course and timing of these changes in the “bridging years” between illness stages. The National Advisory Mental Health Council’s Workgroup Report recommended that in the identification of pathophysiological processes that contribute to symptoms or syndromes “[p]articular attention should be devoted to discovering the sensitive and critical periods when neuroplasticity in specific circuits is greatest and maximally responsive to intervention” [98]. This requires careful phenotyping of neuroprogression throughout the early course of illness and development of predictive models.
Neuroprogression in Early PsychosisAbnormalities of gray and white matter volume, network connectivity, and cognition are well-described in patients with established psychosis, and longitudinal research suggests that neuroprogression may continue well into chronicity in some patients [65,67,99]. However, neuroprogressive changes may begin much earlier in the illness course, some even prior to illness onset. Examination of cognition, gray matter, white matter, and connectivity at various stages of illness suggest that (1) measurable alterations exist in each of these domains and (2) abnormalities do not progress uniformly across stages of illness (Table 1). For instance, cognitive abnormalities appear to be present prior to illness onset in patients with SZ, becoming more widespread by the first episode with profiles qualitatively and perhaps quantitatively similar to chronicity in both BP and SZ [26]. In contrast, while gray matter (GM) and white matter (WM) reductions are reported in multiple frontal, temporal, and parietal regions by the first episode, and these markers appear to become more widespread and pronounced compared to controls into chronicity. Regionally, some brain volume abnormalities appear to be present in first episode at the magnitude seen in chronicity (e.g., hippocampal volume), whereas some structures (e.g., amygdala) that show significant abnormalities in established psychosis show no evidence of abnormalities in high risk [59,100]. Thus, neuroprogression is detectable early in the course of illness; however, neuroprogressive changes across modalities do not occur uniformly.
Neuroprogression and Illness CourseNeuroprogressive changes are related to disease course. Cognitive deficits are predictive of functional disability [3,24], and progressive gray matter loss and increased cerebrospinal fluid volumes are associated with symptom severity, clinical course, and poorer functional outcomes [48,65,101–103]. Associations amongst cognitive and brain measures suggest complex dynamics amongst these domains, and with illness course and functional outcomes [30,58,64,104–106]. For instance, in a recent study functionally connected brain regions “thinned together” in networks related to cognition, showing a dynamic interplay amongst cognitive, structural, and connectivity changes [107].
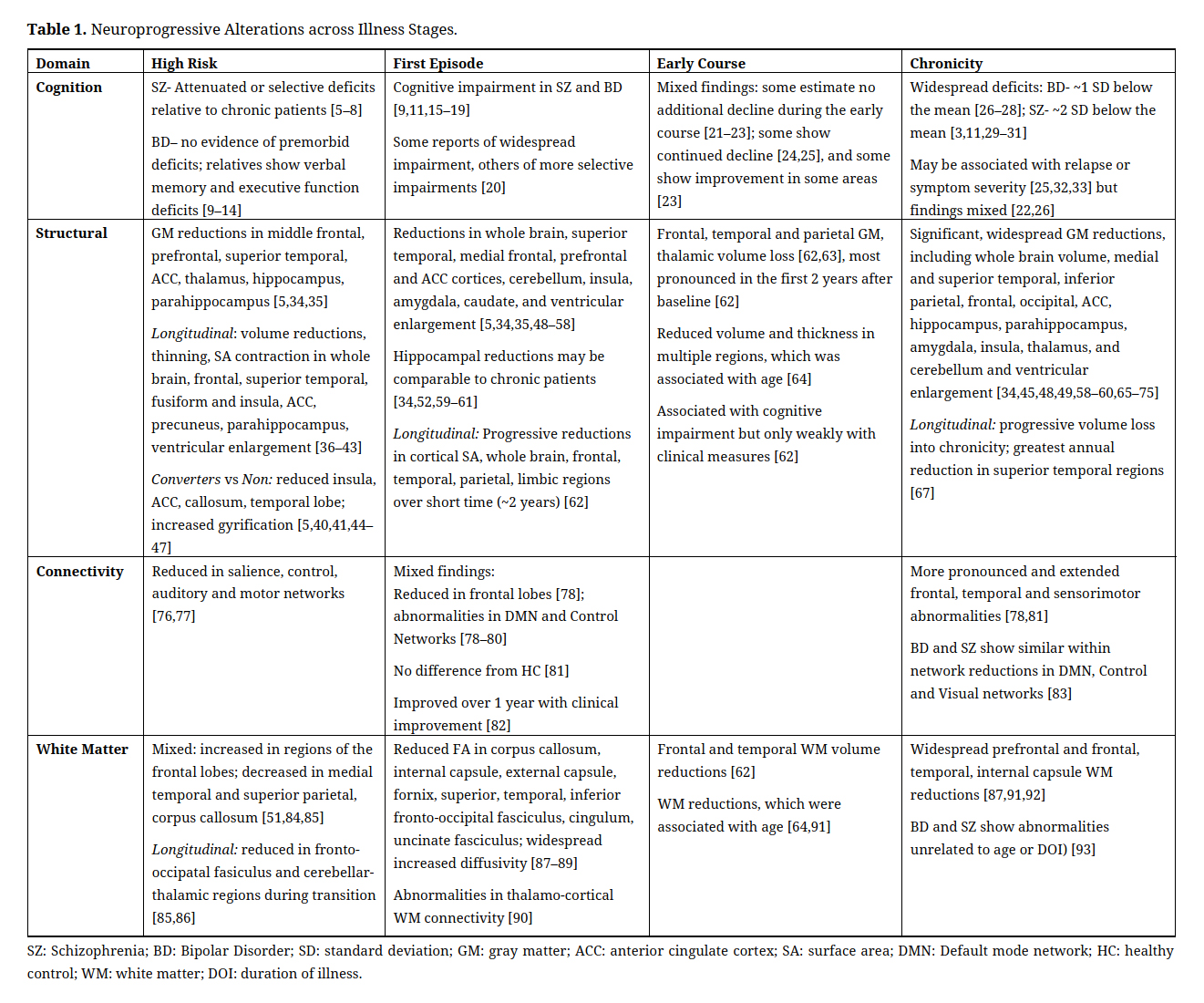 Table 1. Neuroprogressive Alterations across Illness Stages.
Table 1. Neuroprogressive Alterations across Illness Stages.
Together, findings suggest that neuroprogressive changes in the years following psychosis onset occur rapidly in many areas during an important neurodevelopmental window [94], with continued progression throughout the early and mid years. However, the current state of the literature is insufficient to carefully characterize neuroprogressive trajectories at key inflection points [29] and throughout the “bridging” years between onset and established illness—years that are critical for targeted intervention—for several reasons. First, studies of high risk participants often have very low rates of conversion to psychosis resulting in possible “dilution effects” [94,108,109]. Second, most longitudinal studies include only two assessment points forcing the assumption of linearity, and those with more assessments are typically years apart making it difficult to pinpoint inflection points in primary outcomes [94]. It has been suggested that repeated assessments (>2) at relatively short (i.e., at most 1 year apart) intervals are needed to characterize the longitudinal trajectories in each of these neuroprogressive domains [94,110]. Other challenges include considerable methodological variation across studies (e.g., definition of key grouping characteristics (e.g., DOI in first episode studies); inter-scan interval; analysis approach). Additionally, as can been seen in Table 1, relatively few studies focus explicitly on the years between onset and chronicity, despite the critical nature of the early course of illness in terms of prognosis and intervention.
Heterogeneity in Cognition and NeurobiologyHeterogeneity of premorbid adjustment, illness course, and outcomes is the rule rather than the exception in psychotic disorders, (e.g., [62]), and identifiable subsamples may differ in neuroprogressive degree and trajectory (e.g., [111]). Recent reports suggest that abnormalities in brain structure and connectivity are associated with cognitive subtypes in psychosis [112–114]. A recent study found that baseline neurocognitive functioning predicted gray matter reductions in multiple brain regions two years later [115], suggesting that baseline profiles may predict neuroprogressive course. While heterogeneity can interfere with our ability to identify associations and timelines at the group level, it may be possible to leverage this heterogeneity to identify subgroups that share similar behavioral and neurobiological presentation and course toward more individualized prediction and treatment implementation.
Goals and HypothesesEvidence strongly indicates that neuroprogressive changes occur across brain and cognitive measures in patients with psychosis at the time of first episode and throughout the early course of illness. Nonetheless, no studies to date have undertaken the systematic characterization of neuroprogression throughout the early course of illness at a granular level using multimodal assessments in a transdiagnostic sample. Thus, our objective with this project is the systematic, multimodal characterization of neuroprogression through the early years of illness in a cross-diagnostic sample of patients with psychosis. We hypothesize that (1) by interrogating neurocognition, structural brain measures, and network connectivity at multiple assessments across the first eight years of illness we will identify clear neuroprogressive trajectories along our primary outcome variables, and (2) neuroprogressive trajectories will be predictive of clinical and functional outcomes. To accomplish this objective within the project timeline, we will use an accelerated longitudinal design (ALD, described below) modeling multiple neuroprogressive markers by duration of illness, and in association with key clinical and functional measures. A central aspect of this project is that it builds upon the rich neuroimaging, cognitive, and clinical data being collected by the Human Connectome Project in Early Psychosis (HCP-EP; PI: Dr. Martha Shenton). Data collection for this longitudinal study will occur across the Boston HCP-EP sites, and it is estimated that approximately 75% of baseline data for the present project will be drawn from existing data collected in the context of the HCP-EP.
192 patients with DSM-V non-affective (schizophrenia, schizophreniform, schizoaffective, psychosis NOS, delusional disorder, brief psychotic disorder) or affective (major depression with psychosis or bipolar disorder with psychosis) psychosis as determined by SCID-5-RV for DSM-V-RV interview [116] will be enrolled. As noted above, it is expected that approximately 75% of these subjects will have participated previously in the HCP-EP. Patients will be between the ages 18–35 at enrollment, and between the ages of 17 and 30 at the time of their first episode. Duration of illness will be determined via the SCID interview, together with all available collateral data from medical records, treatment providers, and family members. Subjects must have capacity to provide informed consent. Exclusion criteria include MRI contraindication, IQ less than 70 based on medical history or WASI-II [117], DSM-V diagnosis of substance-induced psychosis or psychotic disorder due to medical condition [116] and known brain damage. We will also recruit 36 control participants. Exclusion criteria for control participants include history of DSM-V diagnosis or psychiatric treatment, and all other exclusion criteria noted above. All procedures have been approved by the Partners Healthcare Human Research Committee/IRB, and comply with the regulations set forth by the Declaration of Helsinki.
MaterialsIn order to capitalize on existing data and maximize comparability between data sets, we will use identical materials and procedures to those currently implemented by the HCP-EP. Participants will be reassessed at the same site as their original assessment, including MRI scans.
Behavioral measuresBehavioral measures include the NIH Cognition Toolbox [118], psychosis-relevant HCP Lifespan measures, and additional measures for early psychosis including: (1) Hollingshead Two-Factor scale [119], measure of parental SES; (2) SCID-5-RV in conjunction with medical records and patient/family clinical interviews to confirm diagnosis; (3) The Positive and Negative Syndrome Scale (PANSS) [120]; (4) The Clinical Assessment Interview for Negative Symptoms (CAINS) [121]; (5) The Young Mania Rating Scale (YMRS) [122]; (6) The Montgomery-Asberg Depression Rating Scale (MADRS) [123]; (7) The MIRECC Global Assessment of Functioning (GAF) [124]; (8) HCP-EP Lifetime Medication Record, which assesses past and current medication use; 9) WASI-II Vocabulary and Matrix Reasoning to estimate IQ, and 10) the Seidman Auditory Continuous Performance Test (CPT [125,126]).
Medications represent an important covariate in the study of progressive changes in brain and behavior. We will collect detailed information about mediation use at each assessment, and account for medication effects using analytic models of antipsychotic, Lithium, and total medication load both as moderators and confounders in order to examine the potential role of medications in neuroprogression.
Neuroimaging: MR data acquisition protocolImaging data will be collected on two Siemens MAGNETOM Prisma 3T scanners, one at McLean and one at Brigham and Women’s Hospital. Both use a 32-channel head coil and are actively collecting HCP-EP data using the same sequence employed here. This protocol is similar to the original HCP Lifespan protocol [127], but without the fMRI task, as many subjects with psychotic disorders may not easily tolerate lengthy MR sessions. Total scan time is just over one hour. The scan sequences include: (1) Localizer and Auto Align Scout; (2) Structural T1w (MPRAGE) (0.8 mm isotropic; T1 1000 ms; TR 2400 ms; 208 slices) and T2w (SPACE) (0.8 mm isotropic; TR 3200 ms; 208 slices) (3) Resting state fMRI (rfMRI) of 2mm isotropic; multiband (MB) acceleration × 8; TR 720 ms; acquired twice: once with AP and once with PA phase encoding; 4) Diffusion MRI (dMRI) 1.5 mm isotropic; TR: 3230 ms; TE: 89.20 m; flip angle 78°; MB acceleration × 4, 92 directions in each shell (b = 1500 and 3000) acquired twice: once with AP and once with PA phase encoding. Field maps will be acquired to correct for intensity and geometric distortions.
ProceduresThis project employs an accelerated longitudinal design (ALD) in order to cover the desired timeframe within the constraints of the project. ALDs follow enrollment cohorts longitudinally and thus model both within subject longitudinal and between subject group effects. While most ALDs use age to define cohorts, we will use duration of illness (DOI), which will allow us to estimate DOI-related trends in our primary outcomes. The timing of repeated assessments will be calibrated to each individual’s DOI resulting in measures that span the range from 0 to 88 months after illness onset. We selected a “balanced ALD”, meaning equally spaced measurements across the study, the same number of measurements per cohort, and equal overlap between successive cohorts (in this case, no overlap; see Table 2).
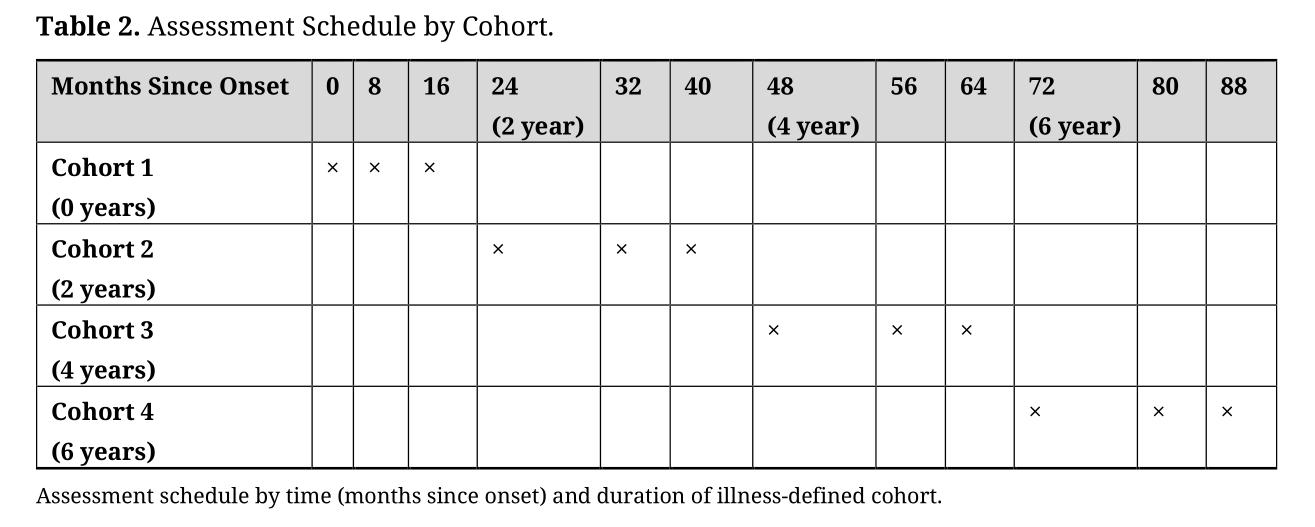 Table 2. Assessment Schedule by Cohort.
Table 2. Assessment Schedule by Cohort.
Assessments will be conducted at baseline, 8-months and 16-months to fully cover the early course of illness (Table 2) at equally-spaced assessments, minimizing overlap of assessment points amongst cohorts (desirable in an ALD [128]). Baseline assessments include MRI scan, clinical and diagnostic interviews, and neuropsychological assessments. Follow up at 8- and 16-months will consist of the MRI scan, clinical and neuropsychological assessments. The follow up interval was selected because scans at least annually have been recommended for assessment of changes that occur relatively rapidly (e.g., [20,94]) as may be the case after psychosis onset [62], and coverage of the time between onset and established illness is needed to fill a critical knowledge gap. Thus, in the context of an ALD, assessments at 8-month intervals permits frequent assessments at multiple assessment points—important for assessment of trajectories without forcing an assumption of linearity [20,94]—while covering nearly 8 years after illness onset. Control participants will also be assessed three times at baseline, 8- and 16-months.
Behavioral assessmentsFollow-up assessments involve approximately 3–3.5 h of behavioral, clinical, cognitive testing. Baseline assessments will also include a clinical diagnostic interview that will add approximately 1.5–2 h. Total assessment time for baseline procedures is expected to take approximately 4.5–5 h over two or more days, within days of the imaging. Participants are provided lunch and/or snacks during the assessment, as appropriate. Reliability was established on all Toolbox measures prior to the start of enrollment. Reliability and consensus diagnosis is ongoing for all diagnostic interview, conducted on a monthly basis with all diagnostic team staff across sites.
MRI scansSubjects complete MRI safety screening prior to scanning. Procedures are described to subjects and they are helped into the scanner by study staff and an MRI tech at the scanning site. Subjects are instructed to remain still during scanning and deformable foam cushioning is used to stabilize the head. Real time image reconstruction and processing are used for quality assurance at the time of scanning. Total scan time is just over 1 h; with MRI safety checks total time at the scan site is approximately 1.5 h.
All MRI data processing and storage are completed via a central database system at Brigham and Women’s hospital, which has been customized to host the project data and to manage daily operations and QC procedures and synchronized to receive data directly from McLean. This upload tool automatically strips all PHI prior to upload, and de-identified data from both sites with QC are stored in this database system. Automated verification of scan acquisition parameters at the time of the scan are followed by a manual review, and a semi-automated QC procedure developed to detect signal drops is run for each scan. Because two different scanning sites will acquire data, special considerations have been taken to ensure that the data quality is homogeneous across sites. Harmonization procedures include Siemens specific QA tools, phantom measurements (fBIRN and NIST phantoms), and traveling human subjects. Scanner reliability was assessed both between scanners and using test-retest assessments within scanners. Intraclass correlations (ICC) were computed on Freesurfer outputs including total GM volume, subcortical GM volume, cortical WM volume, brain segmentation volume, and total ICV, and regionally specific measures based on our primary outcomes. We found ICCs of 0.98–0.99 for broad measures, and 0.90–0.99 for regional measures.
Planned analysesOur primary aim is to map neuroprogression across the first 8 years of illness. We will fit latent growth curve models to the repeated measures of our cognitive and MRI measures on the full sample using techniques that permit the functional form of the trajectories to be determined by the data. Each model will include initial DOI as a covariate to separate between-subject differences from within-subject changes as a function of current DOI at each assessment point [129,130]; the model will also include age at assessment to evaluate the effects of natural aging, which will also be estimated from the control participant data, as well as demographic and clinical covariates. By including the between-subjects and within-subjects separately in the models we are able to determine the extent to which trajectories are more strongly associated with longitudinal change over DOI, or cohort effects. These methods allow examination of inflection points in primary outcome trajectories, and peaks and valleys by DOI. We will also examine diagnostic differences in trajectories.
To evaluate the predictive utility of neuroprogressive change on clinical and community functioning, we will conduct a second set of latent growth curve analyses on clinical and functional measures. We will then build predictive models of our clinical and functional outcomes based on within-subject changes in neuroprogressive markers. We will also explore the possibility that subgroups of subjects demonstrate distinct neuroprogressive trajectories, and differences in clinical and functional outcomes based on these groups using growth mixture models (GMMs). GMMs, an extension of multiple-group growth curve models in which the grouping variable is not specified a priori, can be used to identify subgroups within the data and describe differences in longitudinal trajectories between subsamples. Finally, we will conduct explicit tests of fully dimensional models and combined dimensional and categorical models based on the latent class trajectories groupings.
Determining the timing and course of neuroprogressive changes over the early course of psychosis is essential to the development and implementation of targeted, individualized treatment during a critical time period in which treatments may be maximally effective and the potential for preventing further decline and chronic loss of functioning is at its greatest. Results from this study will (1) hasten the identification of actionable treatment targets that are closely associated with clinical outcomes in order to capitalize on islands of preserved plasticity and maximize their clinical utility, and (2) provide guidance for individualized treatment.
This project includes several key innovations. First, these data will be the first to characterize multiple markers of neuroprogression throughout the early course of psychosis, including inflection points, stabilization points, and associations with clinical course, in a transdiagnostic sample and within a single study paradigm covering the critical years between first episode and established illness. These findings will not only elucidate neuroprogressive processes that are as yet unknown, but will hasten our ability to design treatments in the early course of illness that target actionable mechanisms, potentially preventing further decline and chronic loss of functioning. For instance, if neurocognitive decline predates and predicts structural brain degeneration in associated regions, early treatments targeting cognition for patients with cognitive deficits may improve cognition and halt progression of gray matter loss. Indeed, Eack et al. [131] found that cognitive remediation both improved cognition and slowed gray matter loss in patients with SZ. Additionally, we will examine heterogeneity of neuroprogressive trajectories and their associations with clinical and functional course. The use of an accelerated longitudinal design (ALD) will allow ascertainment of data across a longer timeframe than would be possible in a single cohort longitudinal study, and at more frequent intervals than may be feasible in the same subjects over eight years [128]. To our knowledge this is the first study to employ an ALD based on illness duration in early psychosis. Of course, the use of an ALD rather than a fully longitudinal design in a single cohort introduces the possibility of cohort effects, but we feel that the benefits outweigh the costs by permitting coverage of the full 8 years of interest while reducing the likelihood of large-scale attrition over such a long follow up thereby reducing power especially in the later years, frequent enough inter-assessment intervals to capture inflection and stabilization points in a more fine-grained way, and the potential to fit non-linear models by including 3 assessments per participant. Additionally, we are in a unique position to leverage a major ongoing study of patients in early psychosis to achieve our aims, capitalizing on both the innovations of the HCP-EP and the timing of the study. The HCP-EP is currently enrolling, allowing us to incorporate longitudinal data to the project while data collection is ongoing, and existing data will serve as baseline assessments for a large proportion of the sample allowing us to increase overall enrollment and follow-up and thereby increase power adequately to perform the analyses described above.
KEL developed the project and drafted the original version of the manuscript. SB was responsible for MRI project design, reliability evaluation for imaging data, and drafting of the imaging-based portions of the manuscript. DO was responsible for design consultation and manuscript preparation. MES developed the original project upon which this grant was based, consulted on the design for the present project, and was involved in manuscript preparation.
The authors declare that they have no conflicts of interest associated with this work.
This work was supported by grants from the National Institute of Mental Health to KEL (R01MH117012) and MES (U01MH109977).
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
Lewandowski KE, Bouix S, Ongur D, Shenton ME. Neuroprogression across the Early Course of Psychosis. J Psychiatry Brain Sci. 2020;5:e200002. https://doi.org/10.20900/jpbs.20200002

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




