Location: Home >> Detail
Crop Breed Genet Genom. 2019;1:e190010. https://doi.org/10.20900/cbgg20190010
 ,
Ahmed Jahoor 1,2,
Jeppe Reitan Andersen 1,
Jihad Orabi 1,
Luc Janss 3,
Just Jensen 3
,
Ahmed Jahoor 1,2,
Jeppe Reitan Andersen 1,
Jihad Orabi 1,
Luc Janss 3,
Just Jensen 3
1 Nordic Seed A/S, 8300 Odder, Denmark
2 Department of Plant Breeding, The Swedish University of Agricultural Sciences, 23053 Alnarp, Sweden
3 Department of Molecular Biology and Genetics, Aarhus University, 8830 Tjele, Denmark
* Correspondence: Peter Skov Kristensen.
This article belongs to the Virtual Special Issue "Genetic Gains in Plant Breeding"
Genetically correlated traits can be used for improving predictive abilities of genomic predictions including several traits in multi-trait models. Here, the wheat quality traits thousand-kernel weight, grain protein content, Zeleny sedimentation, and falling number were phenotyped in 1152 advanced winter wheat lines from four cycles of a commercial breeding program. Multi-trait and trait-assisted genomic prediction models including two or four traits were studied and compared with single-trait models. In the trait-assisted genomic predictions, breeding values of the trait of interest were predicted for lines that had been phenotyped for additional traits. Predictive abilities of single-trait models ranged from 0.5 for thousand-kernel weight to 0.65 for falling number based on 10-fold cross-validations. Predictive abilities were in most cases not significantly different between single- and multi-trait models, when no phenotypic data was included for lines in the validation set. However, predictive abilities for grain protein content increased when using trait-assisted models, where the phenotypic data for Zeleny sedimentation or falling number were available for the lines in the validation set. The trait-assisted models also resulted in increased predictive abilities for Zeleny sedimentation, when phenotypic data for grain protein content was included. The latter situation could be relevant for breeding programs for improving wheat quality.
GBLUP, Genomic Best Linear Unbiased Prediction; GEBVs, genomic estimated breeding values; LSO, leave-set-out; MT, multi-trait; ST, single-trait; TA, trait-assisted; TKW, thousand-kernel weight
Genomic prediction is a relatively new technology in plant breeding that can be used for increasing the rate of genetic gain [1]. Genomic prediction has so far mainly been studied using single-trait (ST) models. However, in plant breeding programs, selection of lines is typically based on several traits. Traits might be genetically correlated due to pleiotropy or linkage between QTL influencing the traits. Hence, phenotypic data of correlated traits recorded in breeding programs might be better utilized if they are combined in multi-trait (MT) models for improved prediction accuracies of genomic estimated breeding values (GEBVs) [2].
For many traits, it is challenging to phenotype large numbers of lines at early generations due to shortage of grain quantities. Lines phenotyped for all traits might be used as training set to predict breeding values for the same traits in a breeding set of lines that have not been phenotyped. Alternatively, the lines of the breeding set might also be phenotyped for certain traits and used for prediction of breeding values for unphenotyped traits in the same lines. The latter approach, so-called trait-assisted (TA) genomic selection, significantly increased prediction accuracy of biomass yield in sorghum compared to ST or MT genomic selection [3].
A simulation study showed that predictive accuracies could be increased for a low heritability trait when a correlated, high heritability trait was included in MT models. When the genetic correlation between the traits were increased, the predictive accuracy of the low heritability trait also increased [2]. However, several studies based on empirical wheat data have reported that MT models often did not perform better than ST models [4–6]. In MT models, a large number of parameters has to be estimated, but these estimates might not be sufficiently accurate, when based on the relatively small datasets used in most studies [7]. On the other hand, TA genomic selection could improve predictions compared to ST or MT models. Here, breeding values of the trait of interest were predicted for lines that had been phenotyped for correlated traits [4,6,8].
In the present study, the four quality traits thousand-kernel weight (TKW), grain protein content, Zeleny sedimentation, and falling number were studied. TKW is an important grain yield component that can be correlated with several quality traits [9]. Grain protein content is typically positively correlated with other quality traits, because the majority of wheat grain protein are gluten proteins [10]. Gluten content and quality is essential for baking quality of wheat. When wheat flour is mixed with water, gluten proteins form a network that gives the dough viscoelastic properties and enables it to rise by retaining CO2 that is produced during fermentation [11]. The Zeleny sedimentation test is a relatively quick and cheap method that can be used for estimating the content and strength of gluten in wheat flour [12,13]. Falling number is an indirect measure of α-amylase activity. The α-amylase enzymes break down starch into fermentable sugar. A very low or a very high falling number can both result in inferior bread textures and small loaf volumes [14,15].
The aim of the present study was to evaluate ST, MT, and TA models for genomic prediction of wheat quality traits using advanced lines from a commercial breeding program.
In this study, 1152 F6 winter wheat lines from four different breeding cycles (set2014–set2017) of the plant breeding company Nordic Seed A/S (Holeby, Denmark) were used. The number of lines included from each breeding cycle was 321 in set2014, 314 in set2015, 159 in set2016, and 358 in set2017. A 9.9 m2 plot was grown for each line using standard agricultural practices at Lolland in Denmark. Each line was grown unreplicated in one year at one location.
PhenotypingGrain or flour samples of each line were phenotyped for the quality traits TKW, grain protein content, Zeleny sedimentation, and falling number (Supplementary Table S1) as described in [16]. Briefly, TKW was determined by weighing a small seed sample and counting the number of seeds using image analysis, grain protein content was determined by near-infrared spectroscopy, and Zeleny sedimentation and falling number were determined following the international standard methods (ISO 5529 and ISO 3093, respectively).
GenotypingDNA was extracted from leaves of two-week old seedlings based on a modified CTAB method [17]. TraitGenetics (Gatersleben, Germany) performed the genotyping using the 15K Illumina Infinium iSelect HD Custom Genotyping BeadChip technology [18]. In total, 11,058 SNPs with minor allele frequency of more than 1% and less than 10% missing values were included in the analysis (Supplementary Table S2).
Statistical AnalysisThe following Genomic Best Linear Unbiased Prediction (GBLUP) model was used for ST genomic predictions:
y = Xb + Zu + e (1)
where y is a vector of phenotypes, X and Z are design matrices, b is a vector of fixed effects, u is a vector of additive genetic effects of the lines (u ∼ N(0,Gσ2g), where G is a G-matrix and Gσ2g is additive genetic variance), and e is a vector of residual effects (e ∼ N(0, Iσ2e), where I is an identity matrix and Iσ2e is residual variance). The effects of set and year were confounded and were considered as a fixed effect.
The first method proposed by [19] was used for calculating the G-matrix:
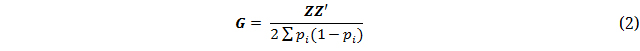
where pi is the minor allele frequency of ith SNP and Z = M − P. M is a matrix with SNP alleles coded as 1, 0, −1, and P is a matrix containing minor allele frequencies of SNP i calculated as 2(pi − 0.5). Missing genotypes were set to 0 in the Z matrix.
The DMU software package was used for REML estimation of variance components and model effects for the GBLUP models [20]. Narrow-sense genomic heritabilities were calculated based on records of single plots:
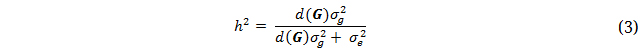
where d(G) is the average of the diagonal elements of the G-matrix, σ2g is additive genetic variance, and σ2e is residual variance.
The following Bayesian SNP-BLUP model was used for genomic predictions:
y = Xb + ZIIuII + e (4)
where ZII is a matrix of SNP alleles coded as 0, 1, 2, and uII is a vector of additive genetic SNP effects.
SNP effects and residual effects were assigned normal prior distributions. SNP effect variance, residual variance and fixed effects were assigned flat prior distributions. Markov Chain Monte Carlo with length of 100,000 cycles including 30,000 burn-in cycles were used for estimation of model parameters. Posterior means were computed using pbayz in the Bayz software package [21], and the CODA package in R was used for checking for convergence to the posterior distribution of all model parameters [22].
Frequentist and Bayesian MT models were used for two different purposes. First, MT models were used to study the genetic correlations between a trait recorded in each of the four breeding sets by considering the records from each set as four different traits. Secondly, MT models and TA models were used for genomic prediction of the quality traits, and two or four of the quality traits were included in the same model. Here the model is shown for two traits:
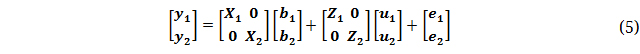
where 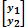 is a vector with phenotypes of trait 1 and trait 2,
is a vector with phenotypes of trait 1 and trait 2, 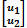 ∼ N(0, G ⊗ H) with variance-covariance matrix H =
∼ N(0, G ⊗ H) with variance-covariance matrix H =  , and
, and 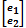 ∼ N(0, I ⊗ R) with residual variance-covariance matrix R =
∼ N(0, I ⊗ R) with residual variance-covariance matrix R =  .
.
Residuals were correlated, when records of the included quality traits were based on the same plot.
When considering traits recorded in sets, residuals were assumed to be uncorrelated.
In the Bayesian MT models, and were assigned normal prior distributions and variance-covariance matrix H and R, respectively.
Two cross-validation strategies were used for evaluating the predictive abilities of the models used for genomic predictions, where one, two, or four of the quality traits were included:
For the 10-fold cross-validations, the wheat lines were randomly divided into 10 folds of equal size. GEBVs of the lines in each fold were predicted using the lines of the remaining folds as training set.
For the leave-set-out (LSO) cross-validations, GEBVs of the lines in each breeding cycle were predicted using lines from the remaining breeding cycles as training set.
The full dataset was used for estimation of variance components in order to obtain as accurate estimates as possible. Predictive abilities were determined as correlation between phenotypes corrected for fixed effects and GEBVs. The theoretical maximum for this correlation is the square root of heritability [23].
In the MT models, the phenotypic data of the lines in the validation set was masked for all traits when predicting GEBVs. In the TA models, the phenotypic data was masked only for one trait in the validation set, and the phenotypic data of the remaining trait(s) were included for all lines.
In total, 1152 winter wheat lines from four different breeding cycles, set2014–set2017, were phenotyped for the quality traits Zeleny sedimentation, grain protein content, TKW, and falling number (Table 1). Estimates of narrow-sense genomic heritabilities, h2, for plot records ranged from 0.51 for grain protein content to 0.58 for falling number.
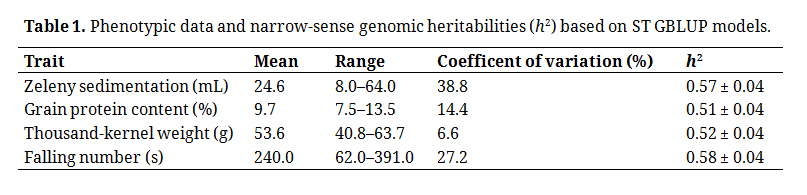 Table 1. Phenotypic data and narrow-sense genomic heritabilities (h2) based on ST GBLUP models.
Table 1. Phenotypic data and narrow-sense genomic heritabilities (h2) based on ST GBLUP models.
The principal component analysis of the G-matrix based on all 1152 lines, showed that the lines of each breeding set was not clearly separated from each other (Figure 1). The first principal component explained 27.4% of the variance, and the second principal component explained 13.7%.
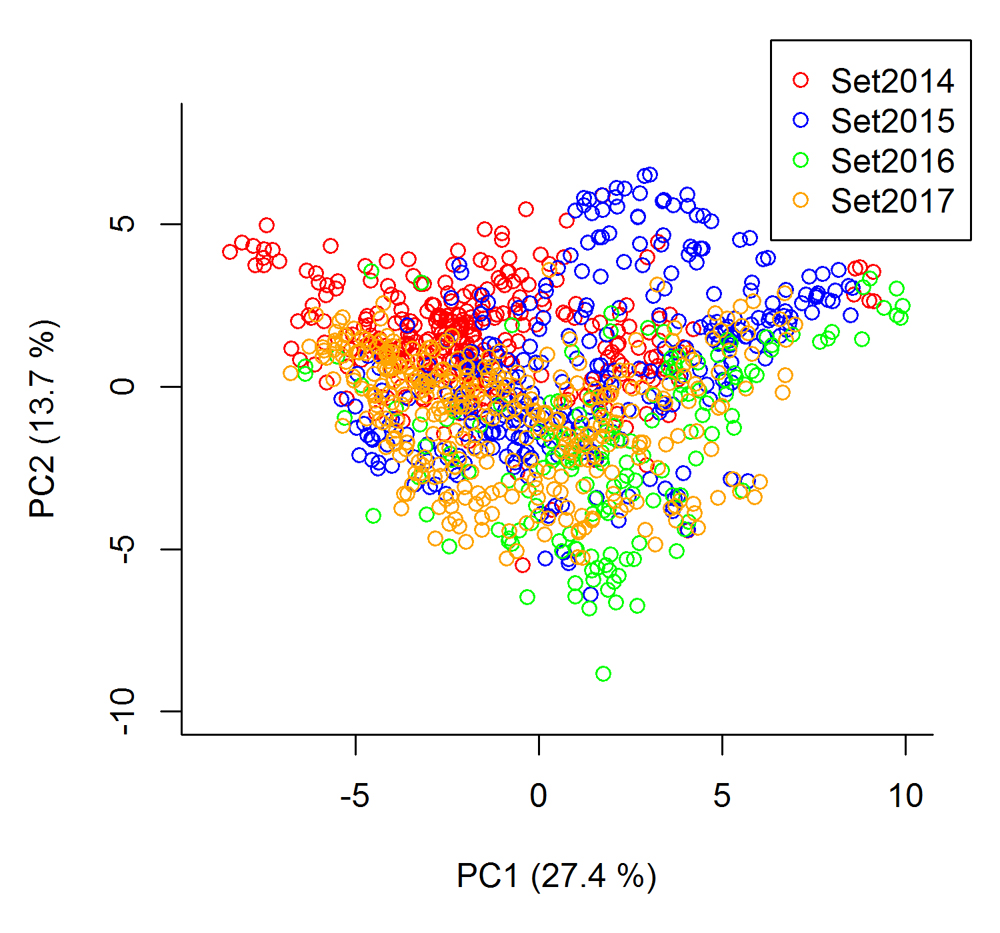 Figure 1. Principal component analysis of the G-matrix based on all 1152 lines from the four breeding cycles, set2014–set2017.
Figure 1. Principal component analysis of the G-matrix based on all 1152 lines from the four breeding cycles, set2014–set2017.
The genetic and environmental correlations between the traits are shown in Table 2. Grain protein content and Zeleny sedimentation were positively correlated both genetically and environmentally. Grain protein content was also genetically correlated with falling number, and the environmental correlation between the two traits was close to zero. Falling number had a negative genetic correlation with TKW and nearly no environmental correlation.
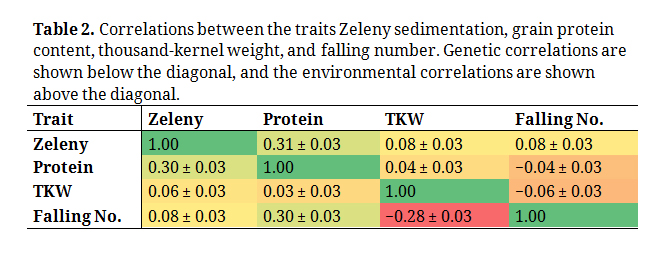 Table 2. Correlations between the traits Zeleny sedimentation, grain protein content, thousand-kernel weight, and falling number. Genetic correlations are shown below the diagonal, and the environmental correlations are shown above the diagonal.
Table 2. Correlations between the traits Zeleny sedimentation, grain protein content, thousand-kernel weight, and falling number. Genetic correlations are shown below the diagonal, and the environmental correlations are shown above the diagonal.
MT models, where a trait recorded in each of the four breeding sets was considered as four different traits, showed that the estimates of the genetic correlations between the traits of each set varied considerably (Table 3). However, the standard errors of the correlations were quite large, especially for grain protein content. For grain protein content and for TKW, the genetic correlations were close to 0 or even slightly negative between some of the sets. For Zeleny sedimentation and for falling number, the genetic correlations were positive between all sets, and the correlations were relativity high in most cases.
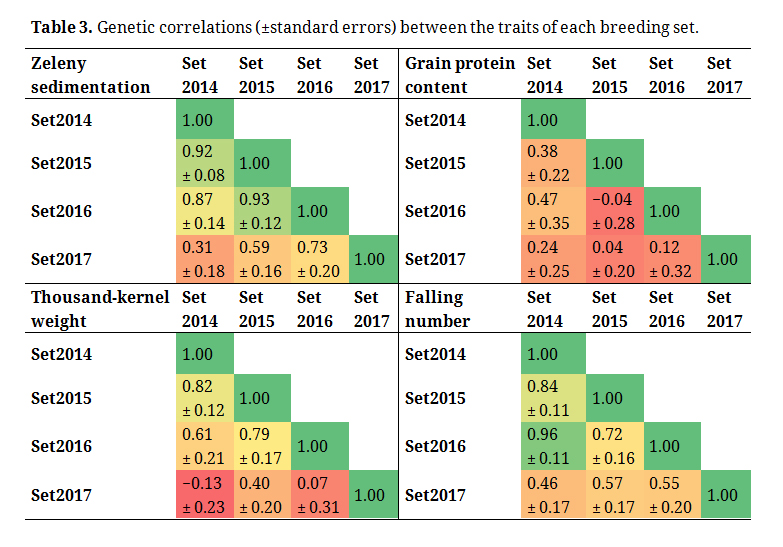 Table 3. Genetic correlations (±standard errors) between the traits of each breeding set.
Table 3. Genetic correlations (±standard errors) between the traits of each breeding set.
For all traits, predictive abilities of ST models were 0.5 or higher based on 10-fold cross-validations (Figure 2). Based on the LSO cross-validations, predictive abilities were lowest for grain protein content (0.13) and highest for Zeleny sedimentation and falling number (0.49). The decrease in predictive ability when comparing the 10-fold and LSO cross-validations were larger for grain protein content and TKW than for Zeleny sedimentation and falling number.
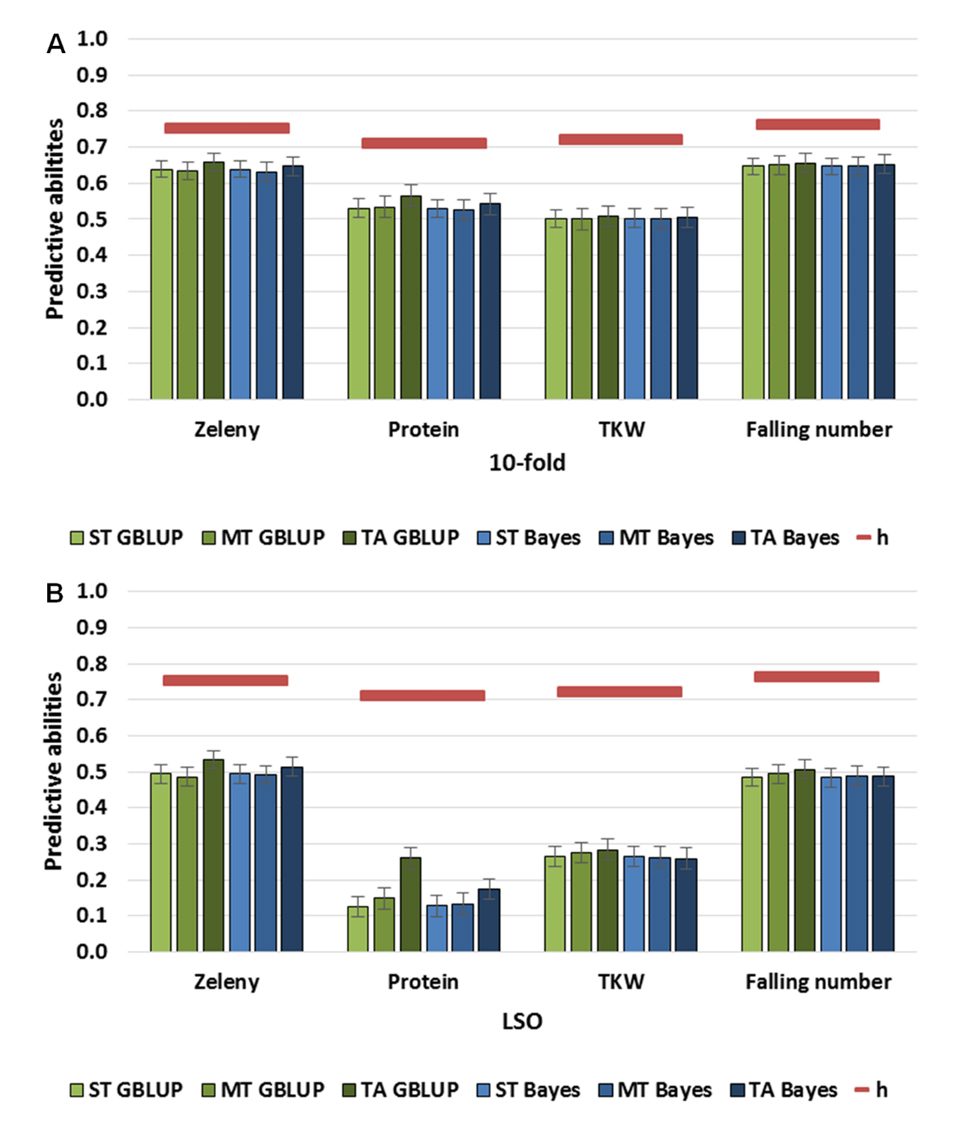 Figure 2. Predictive abilities of single-trait, multi-trait, and trait-assisted GBLUP (green) and Bayesian (blue) models. In the multi-trait models, all four quality traits were included, and the phenotypes were masked in the validation set. In the trait-assisted models, the phenotypes of three traits were known in the validation set, and the fourth trait was masked. The square root of the narrow-sense heritabilities (maximum predictive ability) are shown as red lines. (A) 10-fold cross-validation, (B) Leave-Set-Out cross-validation.
Figure 2. Predictive abilities of single-trait, multi-trait, and trait-assisted GBLUP (green) and Bayesian (blue) models. In the multi-trait models, all four quality traits were included, and the phenotypes were masked in the validation set. In the trait-assisted models, the phenotypes of three traits were known in the validation set, and the fourth trait was masked. The square root of the narrow-sense heritabilities (maximum predictive ability) are shown as red lines. (A) 10-fold cross-validation, (B) Leave-Set-Out cross-validation.
Using GBLUP MT models, where the four quality traits were all included, resulted in similar predictive abilities for all traits compared to the ST models (Figure 2). For grain protein content, the predictive ability was improved when using the TA model based on the LSO cross-validations (0.15 for MT to 0.26 for TA). Predictive abilities of the ST, MT, and TA Bayesian models were all similar to the GBLUP models.
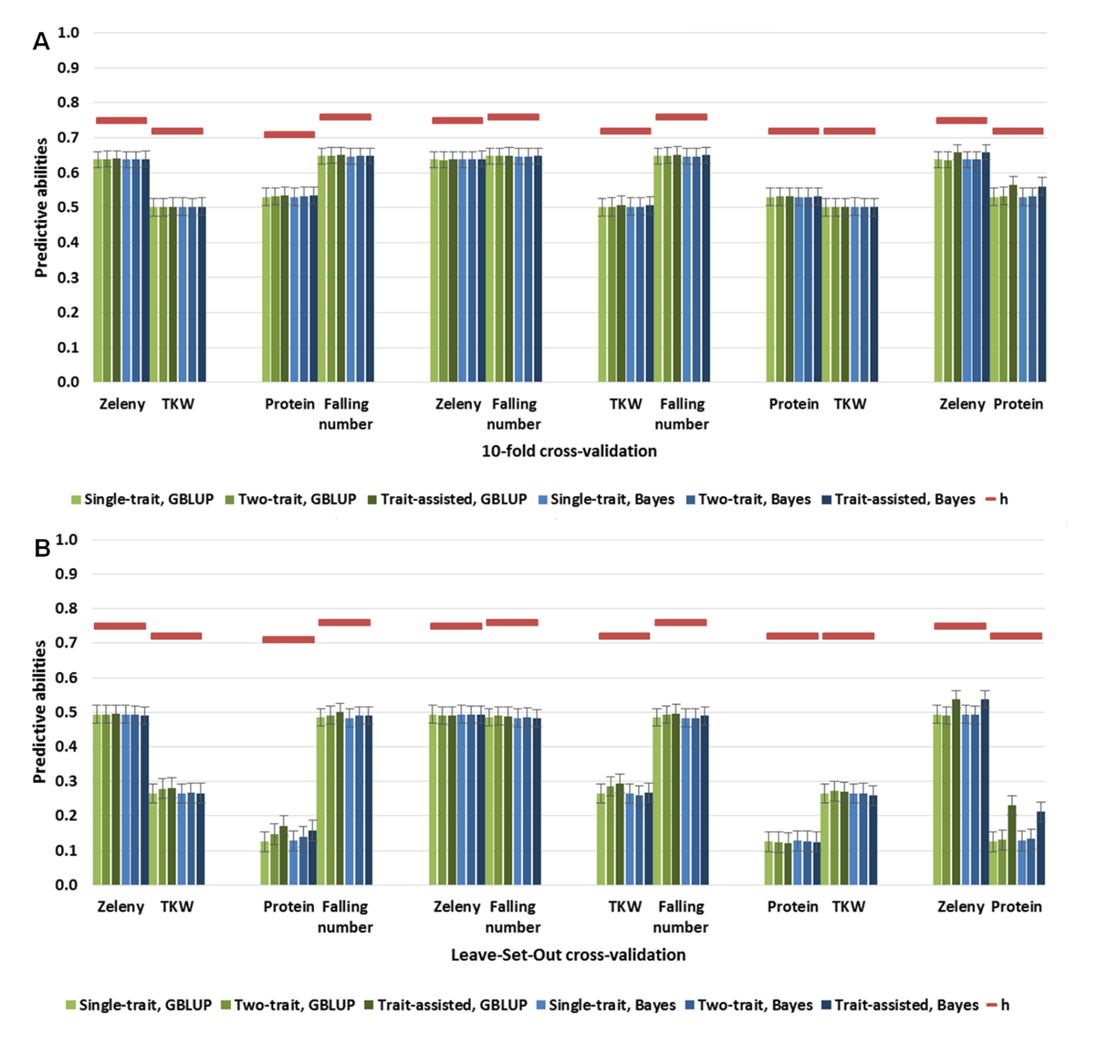 Figure 3. Predictive abilities of single-trait, two-trait, and trait-assisted GBLUP (green) and Bayesian (blue) models. In the two-trait models, the phenotypes of both traits were masked in the validation set. In the trait-assisted models, the phenotypes of one trait were known in the validation set, and the second trait was masked. The square root of the narrow-sense heritabilities (maximum predictive ability) are shown as red lines. (A) 10-fold cross-validation, (B) Leave-Set-Out cross-validation.
Figure 3. Predictive abilities of single-trait, two-trait, and trait-assisted GBLUP (green) and Bayesian (blue) models. In the two-trait models, the phenotypes of both traits were masked in the validation set. In the trait-assisted models, the phenotypes of one trait were known in the validation set, and the second trait was masked. The square root of the narrow-sense heritabilities (maximum predictive ability) are shown as red lines. (A) 10-fold cross-validation, (B) Leave-Set-Out cross-validation.
The four quality traits were also studied pairwise in two-trait models (Figure 3). There were no significance differences in predictive abilities between the two-trait models and the ST models. However, predictive abilities for grain protein content were improved when either falling number or Zeleny sedimentation was included in the TA model (0.13 to 0.17 or 0.23, respectively, based on GBLUP, LSO cross-validations). Furthermore, predictive abilities for Zeleny sedimentation could be improved when grain protein content was included in the TA model compared to the predictive abilities of the ST and MT GBLUP model (0.49 to 0.54, LSO cross-validations).
The narrow sense genomic heritabilities of the four quality traits were lower than in a previous study using fewer lines and fewer breeding cycles [16]. This could indicate that environmental effects and G × E interactions have significant effects on the traits. The gain in predictive ability of MT models compared with ST models is expected to be highest for low heritability traits that are modelled together with traits that have high heritabilities and that are highly genetically correlated [2]. Ideally, the genetic and environmental correlations should be high and have opposite signs in order to have large gains in accuracy from using MT models [7]. Thus, MT models were not advantageous to use for the traits studied here.
The standard errors of the genetic correlations in Table 3 were quite large, so a larger dataset would most likely be needed for more accurate estimates of the correlations, and ideally, the lines should be replicated across years or sets. The lowest genetic correlations were for set2017. This could likely be due to the weather in the summer of 2017. The year 2017 had the wettest summer since 2011, the summer with the fewest hours of sunshine since 2000, and the lowest maximum temperature since 1874 [24]. The results of Table 3 indicate that the traits can be strongly affected by environmental effects and G × E interactions, and underline the importance of multi-environmental trials in breeding programs. Based on data from 32 years of official variety trials in Germany, [10] reported that G × E interactions contributed to the phenotypic variation of quality traits and of grain yield. For grain yield, additive genetics effects accounted only for a small part of the phenotypic variation compared to the quality traits. Predictive abilities for grain yield might be improved when G × E interactions are modelled based on data from multiple environments [25,26]. If phenotypic data from several locations and years are available, outlier environments or breeding cycles could be excluded from the training set in order to increase predictive abilities [27]. However, correct identification of such outliers could be challenging when predicting GEBVs of lines in new breeding cycles.
In agreement with previous studies, predictive abilities of the genomic prediction models were in all cases significantly lower based on LSO cross-validations than based on 10-fold cross-validations (Figures 2 and 3). This could be due to a lower number of lines in the training set, G×E interactions, and reduced genetic relationships between training and validation sets [16]. The decrease in predictive ability when comparing the two cross-validation strategies were largest for grain protein content and for TKW. This is in agreement with the low genetic correlations between the sets for protein content and for TKW and indicates that G × E interactions affected these traits more strongly.
As reported in other studies, MT models did not significantly improve predictive abilities compared to ST models for lines without phenotypic data in the validation set [4–6]. A study based on simulated data showed that MT models could improve predictive abilities for a low heritability trait when including a genetically correlated trait with high heritability. Additionally, the improvements were largest for traits with simple genetic architectures [2]. However, many traits that are relevant for breeding programs have complex genetic architectures. This might explain why the studies using empirical data for wheat lines concluded that MT models did not perform better than ST models. Another reason could be that the variance and covariance components are too inaccurately estimated in the MT models from relatively small datasets [7]. In the present study, the narrow-sense genomic heritability of each of the four traits was higher than 0.5, and there were only small differences in heritabilities between the traits. Furthermore, the highest genetic correlation was 0.30, so the traits were not highly genetically correlated. Thus, MT or TA models could possibly perform better if other traits with higher correlations or larger difference in heritabilities were used, or if data from lines replicated across multiple environments were available [6,26]. Here, GBLUP and Bayesian SNP-BLUP models were used for the genomic predictions and resulted in similar predictive abilities. Other types of models, such as BayesA, BayesCπ or Bayesian Power Lasso, can possibly account for major QTL effects more accurately and therefore, might perform better for traits with simple genetic architectures. However, the predictive abilities of different models are typically quite similar for complex traits [2,16].
A study of Fusarium head blight resistance in hybrid wheat concluded that genomic predictions could be improved if phenotypic data for the correlated traits heading date or plant height was available for predicted lines. ST genomic predictions performed as well as MT predictions when phenotypes of the correlated traits were only available in the training set [28]. In the study of [8], it was suggested to increase the size of training sets for genomic predictions by including lines phenotyped for protein content in order to improve predictions of highly correlated Farinograph or Extensograph quality traits using multi-trait or selection index models. In the present study, predictive abilities for Zeleny sedimentation and for grain protein content were improved when using TA models where the other trait was included. Grain protein content can be phenotyped relatively easily using near-infrared spectroscopy, whereas phenotyping other quality traits, such as Zeleny sedimentation, is more laborious [29]. Thus, all lines that are phenotyped for Zeleny sedimentation in a breeding program would typically also have been phenotyped for grain protein content. Phenotyping a high number of lines for grain protein content in order to increase the predictive ability for Zeleny sedimentation, would therefore be more relevant for breeding programs than vice versa.
High-throughput phenotyping technologies can potentially provide phenotypic for large numbers of lines, which can be included in TA models to improve prediction of traits that are more difficult to phenotype. Such data can either be collected in automated greenhouse facilities or from fields during the growing season using, e.g., drones or tractor mounted systems [4,30,31]. Canopy temperature and vegetation indices can for example be used for improved prediction of wheat grain yield [4,31].
Predictive abilities of ST models were 0.5 or higher for all of the four quality traits based on 10-fold cross-validations. Genomic predictions based on MT models were not significantly more accurate than predictions based on ST models for the studied wheat quality traits. However, genomic predictions of Zeleny sedimentation could be improved using TA models, where the predicted lines had been phenotyped for grain protein content.
PSK performed the data analysis and wrote the draft for the paper. All authors helped designing the study, interpreting results, and revising the paper. Data was acquired by JRA, JO, and PSK. Phenotyping was done by PSK with help from technicians at Nordic Seed A/S. Funding was acquired by AJ, JRA, JJ, and PSK.
This research was performed in a collaboration between Aarhus University and the plant breeding company Nordic Seed A/S. Authors PSK, AJ, JRA, and JO were employed by the company Nordic Seed A/S.
This research was funded by Innovation Fund Denmark, grant number 5139-00018B, Erstatningsfonden for Sædekorn, and Ministry of Environment and Food of Denmark under the Program for Green Development and Demonstrations, grant number 34009-13-0607.
We would like to thank the technicians at Nordic Seed A/S, especially Janni Jørgensen, Eva Szendrei, Jette Andersen, and Hanne Svenstrup, for assisting with field work, DNA extraction, and phenotyping.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
Kristensen PS, Jahoor A, Andersen JR, Orabi J, Janss L, Jensen J. Multi-trait and trait-assisted genomic prediction of winter wheat quality traits using advanced lines from four breeding cycles. Crop Breed Genet Genom. 2019;1:e190010. https://doi.org/10.20900/cbgg20190010

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




