Location: Home >> Detail
Immunometabolism. 2019;1:e190004. https://doi.org/10.20900/immunometab20190004

1 The Research Center for Digestive Tract and Liver Diseases, Tel-Aviv Sourasky Medical Center and Sackler School of Medicine, Tel-Aviv University, Tel-Aviv, 64239, Israel
2 Department of Clinical Microbiology and Immunology, Sackler School of Medicine, Tel-Aviv University, Tel-Aviv, 69978, Israel
* Correspondence: Chen Varol, Tel./Fax: +972-3-6974226.
Incretin peptides, mainly glucagon like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP), are gut derived hormones secreted upon cues from ingested food that regulate glucose concentration, lipid metabolism and gut motility. The classic role of incretins is to convey the status of nutrient availability from the gut to the brain, initiating changes in eating behavior and energy expenditure to maintain body weight. Incretins also act directly on metabolically important peripheral targets in a highly concerted fashion to maintain energy balance and glucose homeostasis. As a result, a myriad of therapeutics for metabolic diseases based on the actions of incretins, particularly GLP-1, are currently under clinical development. Multiple immune cell subsets express receptors for GLP-1 and GIP, but their immunoregulatory roles remain incompletely understood. Innate and adaptive immune cells are intertwined in the homeostatic regulation of central and peripheral metabolism and energy balance, but also contribute to their impairment in the context of metabolic syndrome-related diseases. Hence, delineating the cellular and signaling networks by which incretins operate at the immunometabolic interface has a remarkable translational relevance. In this review, we concisely describe the well-characterized metabolic functions of GLP-1 and GIP, and offer an unprecedented view on the ability of these hormones to modulate immune responses via their direct action on immune cells.
Incretins are gut hormones that facilitate insulin secretion upon food ingestion in a glucose-dependent manner. The glucose-dependent insulinotropic peptide (GIP) and glucagon like peptide-1 (GLP-1) exert their insulinotropic actions on the pancreas via G-protein coupled receptors expressed on islet beta cells [1,2]. Incretins also contribute to the control of energy homeostasis by modulation of satiety and food intake, energy expenditure as well as by additional effects on metabolic organs such as the liver and adipose tissue [2]. Indeed, the last decade is characterized by fast-growing usage of incretin mimetics in the therapeutic armamentarium of type 2 diabetes mellitus (T2DM) and obesity [3,4]. The compelling evidence for the high benefit/low risk profile of GLP-1R agonists may broaden the indications for their utilization in other metabolic diseases, such as cardiovascular diseases and non-alcoholic fatty liver disease (NAFLD). In addition, clinical trials investigating the metabolic effects of combination therapy with GIP analogues are also emerging with overall beneficial synergistic improvement in glycaemic control and body weight [5–7]. However, extending the usage of GLP-1R agonists and implementation of GIPR agonist-based therapies necessitate a better understanding of their mechanisms of action in the central nervous system (CNS), liver and other peripheral tissues. Receptors for GLP-1 and GIP are also found on different immune cell populations, thus suggesting in addition direct immunoregulatory roles for these metabolic hormones. In this review, we aim to provide an up-to-date view of the immunoregulatory properties of these incretin hormones and their potential impact on physiological metabolic actions. To this end, we first describe the recent advances in our comprehension of immune cell networks in adipose tissue and brain, and their concerted modulation of organ-specific metabolic pathways in health and disease. This is followed by a brief description of the key metabolic activities of GLP-1 and GIP. Finally, we summarize accumulating evidence outlining direct anti-inflammatory and protective roles for these incretin hormones in distinct immune cells. There is now reasonable data to suggest that incretin-based therapeutic agents have an anti-inflammatory effect in addition to their ability to regulate blood glucose, which may provide additional benefits in the treatment of T2DM.
Immune Circuits Controlling Metabolism and Energy Balance in Adipose DepotsWhite adipose tissue (WAT) specializes in storing excessive energy in the form of triglycerides (TG) and in liberating fatty acids at times of energy deficiency such as fasting [8]. White adipocytes also release a variety of adipokines and lipid metabolites that signal to other tissues, contributing to systemic energy and glucose homeostasis. These are highly plastic cells that adjust their storage and secretory functions to the nutritional state of the organism [8]. When energy intake exceeds energy expenditure, white adipocytes grow in number (hyperplasia) and in size (hypertrophy) to accommodate energy excess [9]. Visceral WAT is also known for its robust inflammatory response in states of nutrient excess, which contributes to insulin resistance [9]. In contrast to WAT, the major function of brown adipose tissue (BAT) is to dissipate stored chemical energy in the form of heat in response to cold challenge, activation of the sympathetic nervous system or high calorie intake. This is enabled by uncoupling fatty acid oxidation from ATP production via the uncoupling protein-1 (UCP1) [8]. While formerly thought to exist solely in hibernating animals, BAT participates in maintaining energy balance in humans as well, where excess nutrients are dissipated as heat. Brown-like adipocytes termed beige adipocytes were also characterized in human WAT, particularly in subcutaneous WAT (scWAT). These cells arise in response to chronic cold exposure, prolonged β-adrenergic stimulation or high caloric intake [8]. Generally, stress, exercise and cold exposure induce beiging, while pathological processes such as obesity and inflammation reduce it [8]. Of note, in animal models the representative visceral WAT is epidydimal WAT (epiWAT) and the scWAT is inguinal WAT (ingWAT). The WAT cellular ecosystem also harbors a unique blend of immune cells, which cooperate with adipocytes to regulate metabolic homeostasis [10]. During obesity, acute self-limiting inflammation allows healthy expansion of adipose tissue, as it preserves tissue integrity and helps it to adapt to the metabolic needs derived from over-nutrition. However, following prolonged positive energy balance, this physiological response becomes sustained and turns pathogenic. Indeed, obesity is characterized by pro-inflammatory transition of WAT resident and infiltrating immune cells, which contribute to systemic insulin resistance [11]. With the purpose of better understanding the potential immunoregulatory activity of GLP-1 and GIP on fat depot immunity, we will first summarize the current appreciation of their immune cell networks and their support or impairment of metabolism and energy homeostasis.
Macrophages are myeloid immune cells that are strategically positioned in nearly all tissues, where they play essential roles in responses to infection and injury. In addition, each tissue-resident macrophage population exhibits a distinct phenotype linked to the functional specialization of that tissue [12]. In WAT, macrophages are the most abundant immune cells and can comprise up to 40% of all stromal vascular cells in obese adipose tissue [13]. Historically, macrophages were classified as classically activated pro-inflammatory M1 type or as alternatively activated M2 type involved in tissue repair. In light of this polarized classification, it has been suggested that during obesity, WAT-derived excess lipids and inflammatory mediators induce the recruitment of inflammatory Ly6Chi monocytes (correspond to CD14+ monocytes in human) and the shift of adipose tissue macrophage (ATM) polarization towards M1 phenotype [13]. Yet, a more updated dogma grants a metabolically activated phenotype to ATMs that both potentiates inflammation and concomitantly facilitates the removal of dead adipocytes and lipid metabolism [14–16]. The ATM compartment of obese mice and humans is heterogeneous, with CD9+ and Ly6Chi subsets increasing during obesity; the first form crown-like structures near dying adipocytes, are lipid-laden and pro-inflammatory, while the latter are adipogenic [17]. An additional important phagocyte population residing in the WAT are conventional dendritic cells (cDCs) [18]. These are professional antigen presenting cells that bridge innate and adaptive immunity by instructing T cell differentiation and secretion of cytokines and chemokines that modulate immune responses. Early studies in obese mice and patients have attributed a key role to accumulating cDCs in inducing insulin resistance through their activation of Th17 and Th1 pro-inflammatory T cell adaptive immune responses [19–22]. Less is known about their role in homeostatic conditions. In fact, a recent study has shown that WAT cDCs are poised to suppress inflammation by activating the β-catenin and PPARγ pathways, which are also important regulatory mechanisms for fat expansion. Persistent over-nutrition however interferes with these pathways leading to a pro-inflammatory switch of WAT-cDCs [23]. T cells also regulate obesity-induced adipose tissue inflammation. Interferon-γ(IFNγ)-producing CD4+ Th1 cells are increased in obesity, while CD4+ Th2 T cells are decreased [24]. Foxp3+CD4+ regulatory T cells (Tregs) are critical regulators of the inflammatory state of WAT, and are distinct from standard lymphoid tissue Tregs owing to their high fractional tissue representation, high levels of IL-10 and involvement in lipid metabolism. Their unique properties depend on PPARγ [25] and IL-33 [26,27]. Obesity is accompanied by a striking drop of Tregs in WAT [25,28], which may license the pro-inflammatory switch of other immune cells.
The recent past has seen great progress in our comprehension of immune cell circuits controlling energy balance [29]. Studies in mice revealed critical neuron–macrophage interactions, both in the CNS and in peripheral tissues including WAT and BAT, which impinge on energy and metabolic homeostasis (reviewed in [30]). With respect to peripheral neuron–macrophage crosstalk, three independent studies have outlined intimate functional associations between sympathetic neurons and CX3CR1+ macrophages within WAT [31,32] or BAT [33], which were termed sympathetic-neuron associated macrophages (SAMs). Sympathetic innervation of WAT and BAT and production of the catecholamine norepinephrine (NE) is necessary and sufficient for the initiation of lipolysis. In the obese mouse ingWAT, SAM-mediated uptake and degradation of NE seems to contribute to the attenuated NE-stimulated lipolysis characteristic of the obese state [32]. In addition, increased catecholamine catabolism machinery in WAT SAMs, governed by NLRP3-inflammasome, has been linked to lipolysis reduction associated with aging [31]. In BAT, CX3CR1+ macrophages play an important supportive role in local sympathetic innervation [33]. Jointly, these studies highlight the potential immune-metabolic consequences of macrophage-governed tuning of NE availability in adipose tissue.
Type 2 immunity is characterized by production of the specific interleukin cytokines IL-4, IL-5, IL-9 and IL-13, and this immune response is commonly observed in tissues during allergic inflammation, infection with helminth parasites and also during tissue repair. However, type 2 immune cell networks are also an important factor affecting metabolic homeostasis and energy balance. Accordingly, eosinophil-derived IL-4 maintains alternative M2 activation of ATMs to facilitate ingWAT beiging [34,35]. Group 2 innate lymphoid cells (ILC2s) residing in the lean WAT activate eosinophils via IL-5 secretion and polarize ATMs towards M2 phenotype via IL-13 production [36,37]. ILC2s were also shown to directly promote adipocyte beiging via production of Methionine-Enkephalin (MetEnk) peptides that upregulate UCP1 in adipocytes. IL-33 was found to be critical for priming the pro-beiging activity of ILC2s in WAT [36,38]. This type II cytokine also promotes proliferation of ST2 (IL-33 receptor)-expressing Tregs [25,27], and acts directly on adipocytes to promote beiging and thermogenesis thereby offsetting obesity [25,39]. The numbers of WAT ILC2 and eosinophils decline with increasing adiposity, hence licensing obesity-induced inflammatory activation of ATMs [37]. Invariant natural killer T (iNKT) cells are activated by lipid antigens presented by CD1d molecules displayed by adipocytes. Adipose tissue iNKT cells can regulate body weight and restore metabolic homeostasis in obesity [40–42]. They have a unique transcriptional program and produce IL-2 and IL-10 [43]. Adipose iNKT cells are reduced in obesity, but their activation and expansion can lead to potent weight loss via induction of WAT beiging in a pathway mediated by fibroblast growth factor 21 (FGF21) [44]. Nevertheless, other type 2 immune cells may actually favor diet-induced obesity and glucose intolerance. Although mast cell functions have classically been related to allergic responses, recent studies indicate that these cells also contribute to diet-induced obesity and diabetes. Mast cells expand in WAT of obese human and mice. Their genetically induced deficiency or pharmacological stabilization in mice on Western diet reduces body weight gain and levels of inflammatory cytokines, chemokines and proteases, in concert with improved glucose homeostasis and energy expenditure [45]. In light of this imperative crosstalk between metabolism and immune cells within fat depots, there is a great need to identify the mediators that operate at this interface and can couple nutrient signals to the control of inflammation and adaptive thermogenesis. As described ahead, incretins are in this perfect position to modulate immune-metabolic pathways. Nevertheless, despite growing comprehension of the metabolic activity of incretins, their biology in the immune system remains much less clear, plagued by contradictions and unanswered questions.
Immune Circuits Controlling Metabolism and Energy Balance in the CNSThe hypothalamus is a complex CNS structure comprising distinct neuronal populations in addition to other cell types. The hypothalamic arcuate nucleus (ARC) is located at the mediobasal hypothalamus (MBH), next to the third ventricle and the median eminence. In this region, the blood–brain barrier exhibits permissive characteristics, rendering it an arguably preferential location for relaying peripheral signals into neuronal-based CNS metabolic actions. Indeed, ARC is one of the best studied brain regions controlling feeding behavior and energy expenditure, which coordinates peripheral hormonal, nutritional and neuronal signals and also generates a feedback response [46,47]. The ARC contains two types of functionally antagonistic neurons: orexigenic (appetite-stimulating) neuropeptide Y (NPY) and agouti-related peptide (AgRP) expressing AgRP/NPY neurons and anorexigenic (appetite-suppressing) proopiomelanocortin (POMC)-expressing POMC neurons [46,47]. Upon nutrient ingestion, POMC is cleaved to α-melanocyte stimulating hormone, which is released from the POMC axons and binds to melanocortin 3/4 receptors on downstream neurons, leading to inhibition of food intake and enhancement of energy expenditure [48].
Diets rich in saturated fatty acids (SFA) produce inflammation, microgliosis, and neuronal stress in the MBH region. A growing body of evidence pinpoints microglia, the resident macrophages of the CNS, as the cell population that primarily responds to SFA and mediates the inflammatory process in the MBH, which consequently leads to altered neuronal function (e.g., impairment of anorexigenic effect) [30,49]. Remarkably, depleting microglia from the MBH of mice abolishes inflammation and neuronal stress induced by excess SFA consumption and the consequential blunted neuronal response to leptin, therefore reducing food intake [50]. NF-κB-dependent signaling in microglial cells stimulated by dietary excess appears to play a pivotal role in mediating obesity susceptibility via enhancement of food intake and weight gain [51]. Obesity-associated hypothalamic microglia inflammation is biphasic. Initially the resident microglia are affected; however, it is suggested that bone marrow (BM)-derived monocytes gradually replace MBH microglia as diet-induced obesity persists [51]. In this respect, high-fat-diet (HFD) rapidly stimulates expression of the fractalkine chemokine (CX3CL1) in hypothalamic neurons of obese-prone mice. Inhibition of hypothalamic CX3CL1 reduces hypothalamic inflammation and recruitment of BM-derived monocytes that promote the obese phenotype [52]. Yet, the deleterious role of fractalkine in microglia activation during diet-induced obesity has been challenged by another study demonstrating that microglial activation via the CX3CL1-CX3CR1 pathway actually mediates resistance of female mice to an obesogenic diet [53]. While the potential mechanisms underlying hypothalamic microglial activation in HFD-induced obesity remains inconclusive, it is generally accepted that microglia activation plays a pivotal role in the development of hypothalamic inflammation in obesity. Therefore, there is an increased interest in developing pharmacological approaches aiming at dampening their inflammatory activity in the hypothalamus to improve metabolic abnormalities commonly associated with obesity. As described below, incretins that are common in the CNS milieu bear the potential to improve metabolic traits also by restraining microglia activation.
GLP-1 GLP-1—metabolic functionsProduction and signaling: GLP-1 was identified as part of the gene sequence coding for pro-glucagon, which is expressed in L cells in the small and large intestines [1,54]. GLP-1 is produced postprandially by L-cells in the proximal gut. Yet, it is also released at basal levels even during fasting, possibly from colonic L-cells, which are exposed to non-absorbed nutrients, bile acids, and microbial metabolites [55,56]. More recent studies have revealed that L-cells express genes for several other gut peptide hormones in addition to pro-glucagon [57]. In addition to intestinal L cells, pancreatic alpha cells express as well the proglucagon gene and can produce GLP-1 in a process driven by the pancreatic processing proconvertase enzymes. Normal adult alpha cells secrete little amounts of GLP-1, although this secretion is enhanced in diabetes together with the increased expression of the proconvertase enzymes, which can exert an insulinotropic effect on the remaining beta cells [58,59]. GLP-1 is also produced in the CNS, predominantly in the brainstem, and it is transported to various sites in the brain where it can prompt metabolic, cardiovascular, and neuroprotective actions [1]. Circulating GLP-1 is rapidly degraded by dipeptidyl peptidase IV (DPP4), a serine protease that is widely expressed throughout the body, especially in endothelial cells [60]. GLP-1 exerts its effects via the G-coupled protein receptor (GLP-1R), which is present in pancreatic beta and alpha cells, brain, heart and immune cells [1]. The presence of the canonical GLP-1R in hepatocytes is controversial, as it was not detected in mouse hepatocytes, but was described in rat [61] and human hepatocytes [62]. Whether GLP-1R is present in white adipocytes is also a matter of dispute, and as such, many of the effects of GLP-1 on this tissue may be mediated via systemic effects.
Pancreatic effects: GLP-1 and GIP are together responsible for more than 70% of postprandial insulin secretion [63]. GLP-1 has additional uniquely beneficial effects on pancreatic beta-cells; it induces their proliferation and survival [64,65] and attenuates endoplasmatic reticulum (ER) stress via PKA activation, thereby allowing insulin secretion from stressed beta cells [66].
Regulation of food intake and body weight: Extensive evidence has demonstrated that GLP-1R agonism results in profound weight loss [67], and the GLP-1R agonist liraglutide has been approved for obesity treatment [68]. Though numerous mechanisms such as increased thermogenesis and reduced lipid storage in WAT may be involved, human data have clearly shown that reduction in food intake as a result of GLP-1 activity in the CNS accounts for the majority of the weight lost effect [69]. GLP-1R is expressed throughout the brain and can be activated by either locally-produced GLP-1, intestinal L cell-derived GLP-1 crossing the blood-brain barrier or by L-cell-derived GLP-1-goverened stimulation of sensory vagal afferent neurons (VANs) in the gut, thus making it difficult to assess the relative contribution of each pathway [67]. CNS endogenous GLP-1 is considered to play a physiologic role in the long-term regulation of energy balance, while circulating GLP-1 serves as a short-term satiation sign [70]. Different areas and neurons clusters in the CNS were examined as potential targets for CNS- and L-cell- derived GLP-1 anorexigenic effects. Accordingly, endogenous glucagon coding gene (Gcg) and GLP-1R+ neurons with anorexigenic effects upon stimulation have been identified in the brainstem [71,72]. However, the physiologic relevance of this circuit is yet to be fully uncovered. Moreover, the anorexigenic effect of GLP-1 seems to be independent of VAN circuits [73]. Alternatively, studies employing either GLP-1R agonists or silencing of Glpr1 gene expression strongly support a role for both hypothalamic and hindbrain GLP-1R in the physiological control of food intake. The GLP-1R agonist liraglutide can directly activate anorectic POMC/CART neurons in rodents and thus indirectly inhibit orexigenic AgRP/NPY neurons in the ARC to reduce food intake [74]. However, neuronal circuits in the hypothalamus probably account only partially for the anorexigenic response of peripheral GLP-1R agonism [75], and the complete array of GLP-1 agonism response in the CNS is more intricate and demands further investigation.
Regulation of energy expenditure: With respect to thermogenesis, combined GLP-1 mechanisms affect both central and peripheral nervous systems. Very recent studies have demonstrated that administration of liraglutide directly into the hypothalamic ventral nucleus stimulates BAT thermogenesis and adipocyte browning, independently of food intake [76]. However, the opposite effects on thermogenesis and energy expenditure were observed when GLP-1 acted via VANs. Accordingly, specific deletion of GLP-1R in VANs resulted in increased BAT thermogenesis, increased ingWAT beiging and increased energy expenditure, particularly under a HFD regimen [77]. In addition, a population of VANs expressing GLP-1R is synaptically connected to BAT, allowing transmission of GLP-1 signals to BAT [77]. These attenuating effects of GLP-1 on BAT thermogenesis via the gut-to-BAT neuronal pathway suggests that rather than contributing to heat dissipation, postprandial nutrients are allocated to energy conservation, but under nutrient excess, these same mechanisms would lead to obesity. Another possibility is that GLP-1R agonism directly induces brown-like properties in WAT. Indeed, the GLP-1R agonist exendin 4 reduces adiposity by promoting lipolysis, fatty acid oxidation and mitochondrial biogenesis in epiWAT of HFD-fed mice and in vitro in 3T3L1 adipocytes [78]. These effects are dependent on expression of sirtuin 1 (SIRT1), a key regulator of lipid metabolism [78]. Furthermore, liraglutide administration to mice fed with a high-fat-high-sucrose diet, significantly upregulates the expression of brown adipose-specific markers in perigonadal fat in association with upregulation of AMPK-SIRT1-PGC1α cell signaling [79]. Overall, while these studies attribute a pivotal role to GLP-1 in mediating energy expenditure, although the mechanistic pathways are still incompletely understood.
Liver effects: Several clinical studies with GLP-1R agonists have demonstrated its anti-steatotic and even anti-fibrotic effects [80–83]. These beneficial effects were attributed at least in part to GLP-1R agonism-induced-weight reduction, better glycemic control and insulin sensitizing properties [84]. The question whether GLP-1 has direct effects on the liver is still under debate due to uncertainty regarding the presence of the canonical GLP-1R in hepatocytes. Exendin 4 was reported to reduce hepatic steatosis in ob/ob mice and to increase cAMP levels in isolated rat hepatocytes, resulting in reduced lipogenesis, increased β-oxidation and reduced lipid accumulation [85]. Rats deficient for DPP4 and with increased circulating active GLP-1 were also shown to have reduced hepatic lipid accumulation. Moreover, GLP-1 inhibited de novo lipogenesis and increase β-oxidation in isolated rat hepatocytes, a mechanism dependent on AMPK activation [86]. Another study has shown that exendin 4 alleviates hepatic lipid accumulation-induced ER stress in vivo and in vitro via the SIRT1/ heat shock factor 1/Heat shock protein (HSP) pathway [87]. In non-alcoholic steatohepatitis (NASH) patients, liraglutide improved hepatic insulin sensitivity, reduced hepatic de novo lipogenesis and decreased adipose tissue lipolysis [88]. In this study, in vitro treatment of human primary hepatocytes with exendin 4 also inhibited lipogenesis and lipid accumulation [88]. Another study in T2DM patients uncovered that exendin 4 treatment plus pioglitazone reduces hepatic triglyceride content, decreases circulating levels of liver enzymes, and increases adiponectin levels, without changes in body weight, therefore demonstrating that GLP-1R agonists can improve hepatic steatosis independent of weight loss [89]. Collectively, in addition to systemic metabolic effects, direct effects on the liver exerted by GLP-1R agonists may be involved in improvement of steatosis and steatohepatitis.
WAT effects: While the administration of GLP-1R agonists affects WAT, the expression of the canonical GLP-1R on adipocytes is not widely accepted, despite some data from primary human and primate adipocytes indicating the presence of GLP-1R mRNA and protein [90,91]. Central administration of GLP-1 was shown to reduce WAT mass. In addition, central, but not peripheral administration, reduced expression of lipogenic enzymes expression in WAT and their triglyceride content. These effects were shown to be mediated via sympathetic outflow [92]. Compatible with that, infusion of GLP-1 into abdominal subcutaneous fat has failed to increase lipolysis in healthy human volunteers [93]. Altogether, current data suggests that the action of GLP-1 on WAT is predominantly indirect and mediated through augmentation of sympathetic tune.
GLP-1—immunoregulatory functionsOur comprehension of the immunoregulatory features of GLP-1 has greatly benefited from the development of GLP-1R agonists for the treatment of obesity and T2DM. A plethora of studies attributed anti-inflammatory features to GLP-1 in various chronic inflammatory diseases including type 1 and 2 diabetes, atherosclerosis, neurodegenerative disorders, nonalcoholic steatohepatitis, diabetic nephropathy, asthma, and psoriasis (reviewed in [94]). Specifically, 12 weeks of treatment with the GLP-1 agonist exenatide (a synthetic version of exendin 4) in patients with T2DM had significant anti-inflammatory effects, as manifested by reduced production of reactive oxygen species (ROS), inflammatory mediators and NF-κB activation in blood mononuclear cells. Of note was the rapid effect on these markers seen only a few hours after sole exenatide treatment [95]. Similar results were obtained with the DPP4 inhibitor sitagliptin [96]. Hence, these studies outline a possible direct anti-inflammatory effect for GLP-1 agonism on mononuclear immune cells, which is unrelated to weight lost. Below we provide emerging evidence for the direct ability of GLP-1 to modulate the migration and function of distinct innate and adaptive immune cells. In macrophages, GLP-1R agonism seems to be a potent anti-inflammatory stimulator. Indeed, different GLP-1R analogues proved efficient in attenuating LPS-induced pro-inflammatory cytokine and chemokine production and skewing the acquisition of M2- alternatively activated macrophage phenotype in cultures of mouse and human monocyte-derived macrophages [97–101]. In addition, exendin 4 is capable of ameliorating ER stress response and apoptosis in insulin-resistant murine primary macrophages isolated from experimental atherosclerotic lesions [102]. It also potentiates phagocytosis in macrophages and their governed clearance of Listeria monocytogenes [103]. With respect to WAT, adenoviral-guided over-expression of GLP-1 significantly improved insulin sensitivity in diabetic ob/ob mice and this has been associated with its ability to reduce the frequency of ATMs and their governed production of inflammatory cytokines and chemokines [104] (Scheme 1A). In this study, GLP-1R agonist exendin 4 also attenuated LPS-induced inflammation in 3T3-L1 adipocytes [104], though the expression of the canonical GLP-1R by adipocytes is controversial. While these results attribute anti-inflammatory effects to GLP-1 augmentation in WAT ATMs, it remains elusive whether this effect is direct. Concerning microglia, murine BV-2 microglial cells express the proglucagon gene and secrete GLP-1, processes that are both enhanced by cAMP. High levels of the SFA palmitate downregulate proglucagon expression in these cells [105]. GLP-1R is also expressed by BV-2 microglia cells, and its activation counteracts TNFα-induced apoptosis, reduces oxidative stress and induces production of neurotrophic factors and nerve growth factors [106]. In cultured microglia, GLP-1 seems to be produced in activated amoeboid-type microglia and can mediate their morphological transition from non-activated ramified type to activated amoeboid type [107]. GLP-1R agonists also stimulate microglial expression and secretion of β-endorphin in the hippocampus and spinal cord to produce anti-nociceptive effects and neuroprotection [108,109]. Recently, it has been shown in a mouse model of sporadic Parkinson’s disease that the potent brain-penetrant long-acting GLP-1R agonist NLY01 protects against the loss of dopaminergic neurons and behavioral deficits via its direct prevention of microglial-mediated astrocyte neurotoxicity [110]. Therefore, these overall anti-inflammatory neuroprotective effects of GLP-1-microglia interactions may tune down the inflammatory response within the MBH region associated with persistent obesity (Scheme 1B).
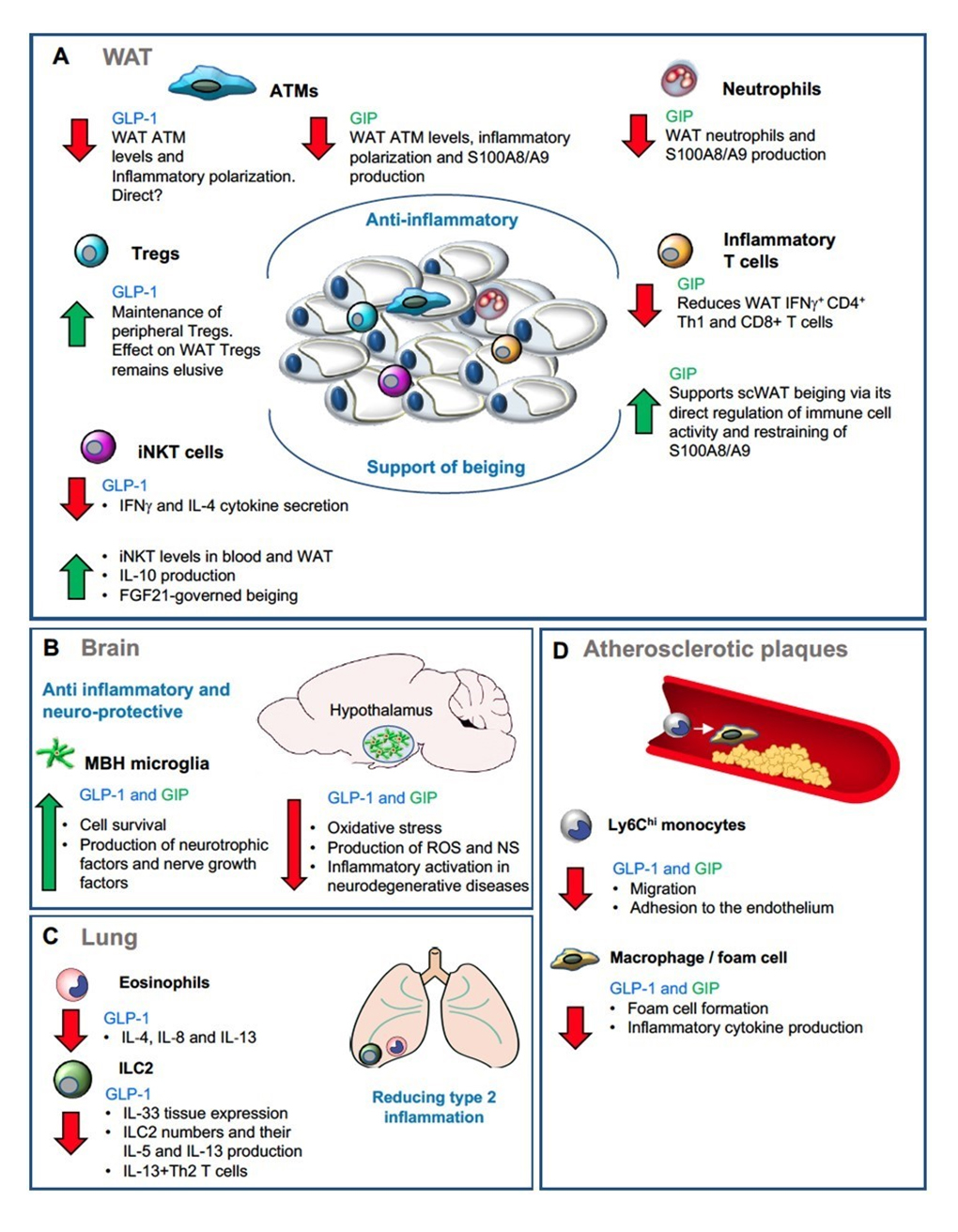 Scheme 1. Site-specific incretin-governed immunoregulation. (A) Schematic illustration of the anti-inflammatory and beiging supportive effects of GLP-1 and GIP in the WAT via their regulation of specific immune cell activities. (B) Schematic illustration of the anti-inflammatory and neuroprotective effects of GLP-1 and GIP in the brain via their regulation of microglia activity. (C) Schematic illustration of GLP-1-governed reduction in type 2 inflmmation in the lung via its supression of eosinophil and ILC2 levels and cytokine production. (D) Schematic illustration of the anti-atherogenic effects of GLP-1 and GIP via their prevention of monocyte infiltration to atherosclerotic plaques and their transformation to foam cells.
Scheme 1. Site-specific incretin-governed immunoregulation. (A) Schematic illustration of the anti-inflammatory and beiging supportive effects of GLP-1 and GIP in the WAT via their regulation of specific immune cell activities. (B) Schematic illustration of the anti-inflammatory and neuroprotective effects of GLP-1 and GIP in the brain via their regulation of microglia activity. (C) Schematic illustration of GLP-1-governed reduction in type 2 inflmmation in the lung via its supression of eosinophil and ILC2 levels and cytokine production. (D) Schematic illustration of the anti-atherogenic effects of GLP-1 and GIP via their prevention of monocyte infiltration to atherosclerotic plaques and their transformation to foam cells.
Obesity and metabolic syndrome are associated with an increased risk of concurrent asthma, and GLP-1 agonists seem to play a prominent beneficial role in these disorders [111]. GLP-1 seems to reduce type 2 inflammation in the lung. Accordingly, GLP-1R is expressed on human eosinophils, and GLP‐1 analogue treatment in eosinophils isolated from mild asthmatic patients significantly decreases their surface expression of activation markers following LPS stimulation and production of IL‐4, IL‐8 and IL‐13, but not IL‐5 [112]. Moreover, GLP-1R agonist reduces lung IL-33 release and the number of ILC2s expressing IL-5 and IL-13 [113]. Similarly, in mice with respiratory syncytial virus (RSV)-induced bronchiolitis, GLP-1R agonist treatment decreases lung expression of IL-13 and IL-33, with concurrent decreases in lung IL-13-producing ILC2, CD4+ Th2 cells, and basophil numbers [114] (Scheme 1C). Given the important roles of type 2 immune cells such as eosinophils [34,35] and the IL-33-ILC2 axis [36,38] in maintaining adipose tissue and energy homeostasis, it will be important to assess the effect of GLP-1 agonists on IL-33 expression and the activity of these cells in WAT. As mentioned above, adipose iNKT cells are reduced in obesity, but their activation and expansion induces potent weight loss in an FGF21 dependent manner [44]. The expression of a functional GLP-1R has been witnessed in human polyclonal iNKT cell lines, and GLP-1 promotes activation of the transcription factor CREB known to regulate expression of anti-inflammatory genes such as Il10 [115]. GLP-1 also induces a dose-dependent inhibition of iNKT cell IFNγ and IL-4 cytokine secretion, but not cytolytic degranulation in vitro [115]. In obese HFD fed mice, short-term treatment with the GLP-1R agonist liraglutide is sufficient to increase iNKT cell frequency in blood and to induce iNKT activation, IL-10 production and proliferation in the WAT [44]. iNKT-induced FGF21 is also augmented by liraglutide [44]. Therefore, these results generate a direct link between GLP-1-iNKT cell interactions and the weight loss effects of liraglutide GLP-1-based therapy (Scheme 1A). Male Glp1r−/− mice exhibit a significantly lower percentage of peripheral Treg cells, suggesting that GLP-1 plays a role in their maintenance [116]. In further support of this, treatment of non-obese diabetic (NOD) mice (a model for type 1 diabetes) with DPP4 inhibitor, leading to sustained GLP-1 and GIP activities, significantly increases the levels of plasma transforming growth factor beta-1 (TGF-β1), an anti-inflammatory cytokine, and increases the fractions of thymic and pancreatic lymph node Tregs contributing to remission of diabetes [117]. Nevertheless, the direct effect of GLP-1 on the levels and function of WAT Tregs remains unexplored (Scheme 1A). With respect to mast cells, which play a deleterious role in obesity-induced insulin resistance [45], liraglutide treatment inhibits their pathological accumulation in experimental models of pulmonary hypertension [118] and obesity-related glomerulopathy [119]. Yet, whether mast cells are a direct target for GLP-1, and what are the effects of GLP-1 agonism on their activity within the WAT, remains unknown. Overall, while these studies attribute direct anti-inflammatory actions for GLP-1 in immune cells, the repertoire of immune cells expressing a functional GLP-1R and the mechanistic impact on metabolic pathways and energy expenditure in homeostasis and disease remain largely elusive.
Atherosclerosis is an arterial disease process characterized by the focal subendothelial accumulation of apolipoprotein-B-containing lipoproteins, immune and vascular wall cells, and extracellular matrix (ECM). Lipoproteins trigger initially endothelial cell activation and subsequently an innate immune response, dominated by Ly6Chi monocytes and macrophages [120–122]. Macrophage foam cell formation, characterized by cholesterol ester accumulation, is the hallmark of early atherogenesis. While both inflammatory macrophages and resolving macrophages can be found in atherosclerotic plaques [122,123], the immunoregulatory mechanisms that dictate macrophage-governed atheroprogression versus atheroregression are incompletely understood [124]. In murine models of atherosclerosis, several studies have indicated the ability of GLP-1 analogues and GLP-1R agonists to induce anti-atherogenic effects by reducing monocyte migration and inflammatory cytokine production and by suppressing their transformation to foam cells [97,100,125]. In vitro, GLP-1 and GLP-1R agonists suppress oxidized-LDL-induced foam cell formation from monocytes isolated from non-obese subjects as well as from THP1 monocytic cell line [125,126] (Scheme 1D). Interestingly however, in monocytes from obese patients, GLP-1R stimulation increases foam cell formation and inflammatory cytokine production in the presence of oxidized-LDL [126], suggesting altered responses to GLP-1 signaling in monocytes under the context of metabolic inflammation.
It is currently appreciated that gut immune cells exert control over systemic metabolism and diet-related metabolic disease by mechanisms that are incompletely understood. In a very recent study, it has been shown that integrin β7+ small intestinal intraepithelial T lymphocytes (natural IELs) modulate enteroendocrine activity by acting as gatekeepers that limit the number of L cells and bioavailability of GLP-1 [127]. Accordingly, integrin β7-knockout mice that lack natural IELs displayed heightened basal energy metabolism and improved glucose tolerance. When fed a high-fat and high-sugar diet, these mice were resistant to obesity, hypercholesterolemia, hypertension, and diabetes. Moreover, β7-integrin blocking protected mice from developing atherosclerosis and led to improved glucose tolerance in a high-cholesterol diet model. Mechanistically, GLP-1 is sequestered by the GLP-1R expressed on IELs, and its loss on β7-integrin-expressing cells leads to increased numbers of GLP-1-producing L-cells and elevated total GLP-1 levels [127]. Therefore, specific targeting of gut IEL-GLP-1R can be considered as a therapeutic approach to mitigate metabolic syndrome diseases. Yet, such an approach would have to consider other results demonstrating that exendin-4 can directly suppress the production of inflammatory cytokines by GLP-1R+ IELs and that Glp1r-/- mice are more susceptible to intestinal injury [128]. Therefore, further studies are needed to assess the metabolic consequences of GLP-1 signaling in IELs. Another layer of immune cell-governed regulation of GLP-1 bioavailability is its enzymatic inactivation by DPP4, which is expressed by multiple gut immune cells [129]. However, it has been shown that DPP4+ cells of BM-derived hematopoietic cell origin mediate the selective degradation of fasting GIP, but not GLP-1 [130].
Further substantiating the immunometabolic crosstalk between GLP-1 and the immune system, GLP-1 secretion by intestinal L cells and pancreatic alpha cells is stimulated by inflammatory signals and cytokines, particularly IL-6, under physiological stressful conditions, such as exercise and endotoxemia [131–133]. This enables maintenance of normal glucose levels under the aforementioned conditions. IL-6 effect on GLP-1 secretion has also been demonstrated in obesity and T2DM [131]. Given the overall anti-inflammatory activity of GLP-1, as mentioned above, inflammation-induced GLP-1 production may be also a feedback mechanism to tune down inflammatory processes.
GIP GIP—metabolic functionsProduction and signaling: GIP is a 42-amino acid peptide hormone produced by K-cells located primarily in the proximal small intestine [55]. GIP release from duodenal K-cells begins upon their sensing of glucose, fat or proteins soon after meal initiation and continues as long as nutrients are released from the stomach [134]. Similarly to GLP-1, GIP is also produced in the CNS, and circulating GIP is also rapidly degraded by DPP4 [60]. GIP exerts its actions by binding to the GIP receptor (GIPR), a G-protein coupled receptor, and its activation results in the stimulation of adenylyl cyclase and Ca2+-independent phospholipase A2, activation of protein kinase A (PKA) and PKB [1].
Pancreatic effects: GIPR signaling activates insulin biosynthesis and secretion in pancreatic beta cells, promotes their proliferation and differentiation and inhibits their apoptosis [135]. GIP-governed insulin secretion is mediated through cAMP/PKA and cAMP/Epac2 pathways [136]. In line with this, Gipr−/− mice display impaired oral, but normal intraperitoneal glucose tolerance [137]. While GLP-1 inhibits, GIP elicits glucagon secretion from alpha cells [134]. Some T2DM patients have been reported to be refractory to GIP-stimulated insulin secretion, though normalization of their hyperglycemia restored sensitivity to GIP-induced insulin secretion [138]. As the molecular basis of this GIPR resistance is incompletely understood, GLP-1 has become a parent compound of incretin‐based glucose‐lowering medications.
Regulation of food intake: With respect to GIP effects in the CNS, there are no conclusive effects on food intake [97]. However, GIP stimulates neural progenitor cell proliferation in the hippocampus and has been demonstrated to have protective effects and improve cognitive function in several neurodegenerative diseases such as Alzheimer and Parkinson [139].
WAT effects: Several studies have underscored a role of GIP as an anabolic hormone in adipose tissue, where it increases adipose tissue mass under nutrient excess. Accordingly, treatment with GIP induces adipocyte differentiation in vitro, and together with insulin, it increases fat accumulation [140,141]. Moreover, GIP increases lipoprotein lipase (LPL) activity, TG uptake and storage in white adipocytes, a process dependent on insulin presence [142]. GIP also reduces lipolysis and free fatty acid secretion in adipose tissue via transcriptional reduction of 11β-hydroxysteroid dehydrogenase type 1 [143]. Some of the main in vivo conclusions about the role of GIP as an obesogenic hormone have come from studies with Gipr−/− mice or administration of DPP4-resistant GIPR agonists. Studies with Gipr−/− mice indicating their resistance to HFD-induced obesity have attributed obesogenic characteristics to this hormone [144]. However, the reduced insulin secretion in these mice may also explain their failure to accumulate adipose tissue mass. This notion is also supported by the fact that mice with GIPR conditional deletion in pancreatic beta cells also fail to gain weight under HFD, despite the fact they have functional GIPR in adipocytes [145]. One study showed that in vivo administration of DPP4-resistant GIPR agonist [D-Ala2]GIP to mice receiving HFD increased insulin resistance [146]. However, our own studies with chronic long-term administration of [D-Ala2]GIP have demonstrated improved insulin resistance, accompanied by markers of increased fat storage in adipocytes, such as adipocyte hypertrophy and increased expression of lipid droplet proteins [147]. We suggest that GIP-induced TG storage capacity in white adipocytes is a protective mechanism under nutrient excess, since WAT is the organ best suited for fat storage, unlike liver or muscle where TG storage causes insulin resistance. In addition, as described below, [D-Ala2]GIP also mediates significant anti-inflammatory effects in WAT (Scheme 1A), which may contribute as well to its-governed improved metabolic phenotype.
Bone effects: Previous studies demonstrated the existence of an entero–osseous axis and established a major role for incretins in the nutrient-dependent regulation of bone metabolism. GIPR is present in both bone forming osteoblasts and bone-resorbing osteoclasts, yet GIP is considered an anabolic hormone favoring bone formation [148,149]. In cultured osteoblastic cell lines, GIP treatment induces proliferation, differentiation and mineralization and inhibits apoptosis [150,151]. Studies with two existing types of GIPR-deficient mice have shown dissimilar results. Mice with exon 4–5 deletion had decreased bone formation parameters, as determined by bone mineral density and content, trabecular bone volume and increased resorption parameters [152]. Mice with exon 1–6 deletion had reduced bone strength and cortical thickness, despite increased number of osteoblasts and increased bone resorption with higher numbers of osteoclasts, suggesting increased bone turnover [153]. Despite these differences, other studies further indicate that GIP favors bone formation over resorption. Accordingly, mice overexpressing GIP display significant increases in bone mass [154]. Moreover, in mice with Streptozotocin-induced type 1 diabetes mellitus, [D-Ala2]GIP treatment was sufficient to significantly prevent deterioration in bone matrix-quality associated with this model [155]. In human patients, short-term treatment with GIP has no effects on bone resorption [156]. However, a loss-of function polymorphism in GIPR is associated with a strong increase in fracture risks in women [157]. Despite contradictory findings, GIPR agonists may prove useful in the treatment of osteoporosis and fragile bone syndrome associated with diabetes [156].
GIP—immunoregulatory functionsAs mentioned above, accumulating data reveals a controversy concerning the role of GIP in the control of body weight. Similar controversy surrounds the role of GIP in obesity-associated inflammatory responses. GIP administration in leptin-receptor deficient db/db mice increased ATM infiltration accompanied by higher levels of CCL2 and IL-6 mRNA expression [158]. Comparable results were also obtained by acute administration of GIP in healthy slightly obese volunteers [159]. In contrast, HFD-fed mice with enhanced genetic expression of GIP exhibited reduced ATM infiltration and pro-inflammatory cytokine expression and an ameliorated metabolic phenotype [160]. In further support of this, we have shown that continuous administration of the long-acting GIP analogue [D-Ala2]GIP reduces inflammation in the epiWAT of HFD-fed mice, as manifested by fewer infiltrating innate and adaptive pro-inflammatory immune cells, attenuated production of pro-inflammatory mediators and improved insulin sensitivity [147] (Scheme 1A). These opposing results likely reflect the integrated pleiotropic functions of GIP in different tissues and cell types. Indeed, we have recently discovered a direct immunoregulatory role for GIP in controlling inflammation and body weight [161]. In contrast to the overall metabolic protection of mice with germline global inactivation of GIPR fed with HFD [144], BM chimera mice with immune cell-targeted GIPR-deficiency displayed greater weight gain (despite similar food intake), insulin resistance, hepatic steatosis and significant myelopoiesis concomitant with impaired energy expenditure and ingWAT beiging [161]. Further conditional targeting of GIPR-deficiency to myeloid immune cells pinpointed a mechanistic role for GIP in restraining the deleterious effects of myeloid cell-derived alarmin S100A8/A9 (calprotectin) on ingWAT beiging [161]. A direct anti-inflammatory role for GIP in myeloid cells has also been described in the context of atherosclerosis (Scheme 1D). Accordingly, continuous administration of GIP in diabetic and non-diabetic apolipoprotein E-null (Apoe−/−) mice suppresses atherosclerotic lesions, macrophage infiltration and foam cell formation in the aortic wall [162,163]. Complementary in vitro experiments have shown that GIP treatment of peritoneal macrophages significantly reduces foam cell formation [163]. Moreover, viral over-expression of GIP in Apoe−/− mice on Western diet reduced atherosclerotic plaque formation, macrophage infiltration and improved plaque stability [164]. in vitro, GIP treatment was sufficient to reduce MCP-1-induced monocyte migration as well as LPS-induced IL-6 secretion and MMP-9 activity in macrophage cell line. This was attributable to GIP-dependent inhibition of NF-κB, JNK, ERK, and p38 in the endotoxin activated macrophages [164]. The gene encoding for GIPR (Gipr) has also been found in human circulating and gut cDC subsets [165–167], but the physiological relevance remains unexplored. With respect to microglia, functional GIPR has been detected in primary murine microglia cells. GIP treatment upregulates in microglia cell lines the secretion of neurotrophic growth factors and antioxidant enzymes. It also inhibits microglia death and the secretion of reactive oxygen and nitrogen species [106]. Further substantiating the therapeutic potential of GIP-microglia axis, GIP analogs reduced neuro-inflammation by lowering microglial activation in models of Alzheimer’s [168,169] and Parkinson’s [170,171] diseases (Scheme 1B). Altogether, these studies suggest anti-inflammatory and immune protective roles for GIP in macrophage type of immune cells in fat depots, atherosclerotic plaques and the brain, all sites with critical influence on metabolism.
The BM contains controlled specialized microenvironments, or niches, that regulate the quiescence, proliferation, and differentiation of hematopoietic stem and progenitor cells (HSPC). Given the expression of GIPR in cells constituting the BM HSPC niche and the altered architecture and matrix of Gipr−/− bones, we endeavored to study the regulation of BM hematopoiesis by GIP [172]. Indeed, we have shown that GIP supports BM hematopoiesis through its regulation of the hematopoiesis-supportive activity of stromal cells composing the BM HSPC niche [172], therefore suggesting another layer by which GIP may modulate immune responses. Datamining into gene expression databases revealed fairly comparable distributions of Gipr expression in various hematopoietic cell types at distinct anatomical sites (Varol and Fishman, unpublished observations). Therefore, there is great importance in determining the biological importance of GIPR in distinct immune cells, specifically in the context of metabolic inflammation and atherosclerosis. Such studies would be essential for understanding the safety of the emerging GIPR-based therapeutics (both agonists and antagonists) [3,7,173].
While it is well-established that inflammation increases GLP-1 levels, how inflammatory stimuli mediate GIP secretion remains largely unknown. In one paper though, increased GIP secretion has been demonstrated in response to endotoxin, and this was driven by IL-1β [174]. In contrast to the inducible effect of IL-6 on GLP-1 secretion [131,132], circulating GIP levels are significantly increased only in response to IL-1β, but not IL-6 or TNFα administration [174], suggesting that GLP-1 and GIP production is controlled by different inflammatory pathways.
Despite the recent advance in deciphering the metabolic functions of incretin hormones, their effects on the immune system have been mostly overlooked. Immune cells function as integral parts of their host tissues during homeostasis, but also play critical roles in the initiation and propagation of pathophysiological pathways during disease. This is profoundly evident during obesity, when pathological immune cell activities at sites such as the brain and WAT alter homeostatic metabolic pathways that are in charge of controlling body weight, glucose concentration and energy expenditure. Accumulating studies illuminate direct immunoregulatory effects for GLP-1 and GIP on different immune cells, usually with anti-inflammatory and protective nature, though this is still a matter of dispute, especially with regards to GIP. Paradoxically indeed, both GIPR agonism and GIPR antagonism either attenuate or augment inflammation [7]. As a result, pharmaceutical strategies enabling either activation [175,176] or antagonism [177] of GIPR signaling are being simultaneously pursued for the treatment of diabetes and obesity. Therefore, there is a great unmet need to dissect the exact repertoire of immune cells capable of sensing these incretins, their immune responses and the resulting impact on central and peripheral metabolism. However, determination of cells expressing a functional GLP-1R or GIPR remains so far challenging mainly due to the uncertain sensitivity and specificity of commonly used antisera [90]. While single cell or bulk comparative gene expression profiling may be sensitive enough to illuminate potential immune cell subsets expressing these receptors, further direct functional assays examining ex vivo in a cell specific manner GLP-1/GIP signaling effects will be complementarily important. Adding to this complexity, many of the immunological studies on GLP-1 and GIP were performed with cell lines or prototype examples of the different immune cells. Given the complex and unique signature, organ specialization and networking of immune cells at sites such as the brain, liver and WAT, efforts should be taken to explore the immune cell specific-incretin effects within their native environments. This would necessitate for example transgenic models allowing the targeting of GLP-1R or GIPR deficiencies to specific immune cell subsets. In another manner, GLP-1 and GIP operate at spatial and temporal overlapping manners and share similar signaling machinery. It will be important to pinpoint the task division and crosstalk between these incretin hormones. Finally, EECs release more than 20 different hormones in response to a variety of stimuli, all with profound impact on metabolism [178]; some possess also substantial immunoregulatory properties, such as serotonin [179,180] and ghrelin [181]. Therefore, further studies are needed to explore the interplay between these hormones and their concerted immunometabolic functions.
Altogether, it is reasonable to assume that incretins may exert at least a part of their beneficial metabolic effects by regulation of innate and adaptive immune responses in the different tissues. Deciphering this innovate link between the incretin and immunity will extend our understanding of how sustained food ingestion upregulates hormonal signals from the gut, which in turn provides unique information to different tissues, thereby contributing to control of nutrient-related inflammation and energy homeostasis.
SF, IZ and CV wrote the paper with input from all authors.
The authors declare that they have no conflicts of interest.
This research was funded by the Israel Science Foundation, grant number [1146/16].
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
Fishman S, Zvibel I, Varol C. Incretin Hormones in the Control of Immunometabolism. Immunometabolism. 2019;1:e190004. https://doi.org/10.20900/immunometab20190004

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




