Location: Home >> Detail
Immunometabolism. 2019;1:e190007. https://doi.org/10.20900/immunometab20190007
 ,
Dimitrios Mougiakakos
,
Dimitrios Mougiakakos
Department of Internal Medicine 5—Hematology and Clinical Oncology, University Hospital Erlangen, Erlangen, 91054 Germany
* Correspondence: Martin Böttcher, Tel.: +49-9131-8543187.
This article belongs to the Virtual Special Issue "T Cell Metabolic Reprogramming"
Since the discovery of gain-of-function mutations in the tricarboxylic acid (TCA) cycle enzyme isocitrate dehydrogenase (IDH) and the resulting accumulation of the metabolite D-2-hydroxyglutarate (D-2HG) in several tumor entities (such as glioma, acute myeloid leukemia (AML), and cholangiocarcinoma) about 10 years ago research has focused on the tumor cell-intrinsic consequences. D-2HG acting as an oncometabolite was shown to promote proliferation, anoikis, tumorigenesis, and differentiation block of hematopoietic cells in an autocrine fashion. Although the prognostic value of the different types of IDH mutations remains controversial the development of inhibitors against mutated IDH is flourishing. On the other hand, serum levels of D-2HG proved to be a more robust adverse prognostic marker in AML and glioma. Surprisingly, until recently only few studies on the paracrine effects of this oncometabolite on the tumor microenvironment with particular focus on the innate or adaptive immunity were available.
Now, three recent publications focused on the paracrine effects of tumor-derived D-2HG on T-cells in the context of AML and glioma. It was shown that T-cells are capable of efficiently taking up D-2HG in vitro, which was mirrored by 2HG-enriched T-cells exclusively found in samples from patients with IDH-mutated AML and glioma. Furthermore, all three studies describe an impairment of T-cell activation (although to different extents). The published effects could be at least partly attributed to metabolic alterations evoked by D-2HG influencing amongst others mTOR signaling, Hif-1α protein stability, the balance between aerobic glycolysis and oxidative phosphorylation, and the abundance of ATP (with according changes of AMPK activation). In the context of glioma it was further shown that IDH mutations and high D-2HG levels lead to reduced T-cell migration and consequently lowered T-cell infiltration at the tumor site. Moreover, two of the studies showed an increased frequency of FoxP3+ Tregs. Nevertheless, effects on downstream mechanisms and consequences have been differently addressed in the independent studies, and taken together the findings shed more light on the potentially targetable sites for improving therapeutic approaches. While the work by our group demonstrated profound impairments of Th17 polarization resulting from D-2HG-triggered Hif-1α protein destabilization the comprehensive analyses by Bunse et al. highlighted an inhibitory effect of D-2HG on the intracellular calcium signaling (downstream of the T-cell receptor) and an activation of AMPK signaling with resulting NFAT inhibition leading to the aforementioned activation defects.
The summarized results of all three studies emphasized the importance of D-2HG not only as an autocrine but also a paracrine oncometabolite capable of shaping the tumor microenvironment in several facets adding to the common concept of tumor immune escape mechanisms. Those findings could lead to further improvements of the current targeted treatment strategies applied to patients harboring IDH mutations especially in view of the increasing importance of (combined) immune-based therapeutic approaches.
Alpha-hydroxyglutaric acid (2-hydroxyglutarate, 2HG) has emerged as an important oncometabolite in the past years. Monoallelic point mutations in the enzyme isocitrate dehydrogenase (IDH) occur amongst others and most frequently in gliomas [1], glioblastomas [2], and acute myeloid leukemia [3]. Those mutations which occur in the cytosolic IDH1 and the mitochondrial IDH2 isoform lead to a loss-of-function in terms of isocitrate conversion to α-ketoglutarate (αKG). Importantly, they also lead to a gain-of-function reducing αKG to the d-enantiomer of 2HG (D-2HG) [4,5](Figure 1). Numerous studies could demonstrate autocrine effects of D-2HG on the tumor cells through involvement in tumorigenesis/leukemogenesis and tumor progression. Acting as a competitive inhibitor of αKG-dependent dioxygenases it blocks the activity of histone and DNA demethylases as well as the TET family of 5mC hydroxylases [6,7]. Together with the inhibition of AlkB proteins prohibiting DNA damage repair mechanisms [8] this leads to profound alterations in the genetic and epigenetic landscape. While some studies demonstrate an inhibitory effect of D-2HG on prolyl hydroxylases leading to e.g., impaired collagen maturation [9] the prolyl hydroxylase PHD2 (fostering the proteasomal degradation of Hif-1α) was found to be activated by D-2HG [10]. Furthermore, IDH mutations and D-2HG have been reported to change the intracellular redox milieu by different mechanisms including alterations in the NADP/NADPH ratio [11] and depletion of glutathione [12]. Taken together, the complex interactional pattern aiding in proliferation and anoikis as well as differentiation block that enable tumorigenesis, progression, and dissemination are already quite well understood. Although the prognostic value of different IDH mutations in the distinct tumor entities is still a matter of debate, several inhibitors of mutant IDH are undergoing clinical trials [13–15] or have already been approved by the FDA such as Ivosidenib and Enasidenib for AML [16,17]. The occurrences and significances of IDH mutations with the consequential production and accumulation of D-2HG, as well as their resulting effects in the context of different tumor entities have been comprehensively reviewed elsewhere [18–20].
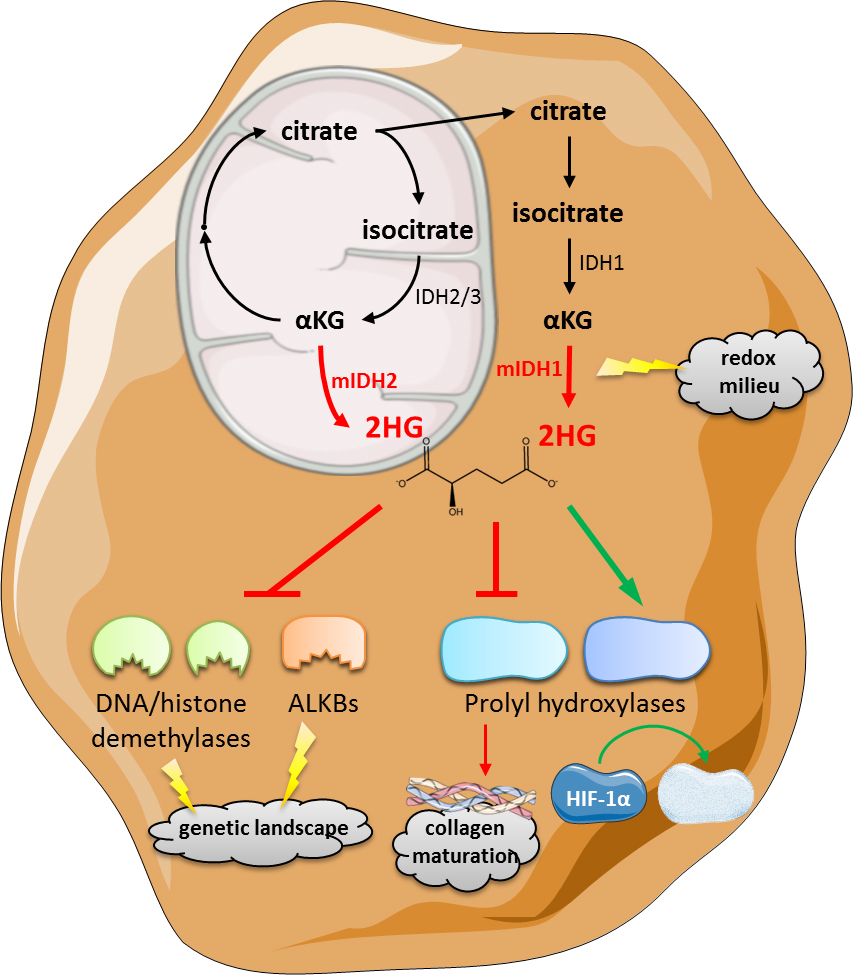 Figure 1. Mechanisms of mutant IDH/D-2HG associated tumorigenesis/leukemogenesis. D-2HG is produced by neomorphic mutant IDH1 (cytosolic) or IDH2 (mitochondrial) through reduction of αKG thereby consuming NADPH. Together with depletion of GSH this leads to changes in the intracellular redox equilibrium. Newly produced D-2HG further inhibits αKG-dependent enzymes including DNA and histone demethylases, and the TET family of 5mC hydroxylases as well as AlkB proteins leading to alterations of the genetic/epigenetic landscape. While prolyl hydroxylases involved in collagen maturation are inhibited, PHD2 that fosters proteasomal degradation of Hif-1α is activated. Consequently, D-2HG is capable of inducing and promoting tumorigenesis and tumor progression. For further details see reviews [18–20]. Mitochondrial IDH3 converts isocitrate to αKG in a NADH-dependent manner. Mutations in the IDH3 isoform have not been reported yet.
Figure 1. Mechanisms of mutant IDH/D-2HG associated tumorigenesis/leukemogenesis. D-2HG is produced by neomorphic mutant IDH1 (cytosolic) or IDH2 (mitochondrial) through reduction of αKG thereby consuming NADPH. Together with depletion of GSH this leads to changes in the intracellular redox equilibrium. Newly produced D-2HG further inhibits αKG-dependent enzymes including DNA and histone demethylases, and the TET family of 5mC hydroxylases as well as AlkB proteins leading to alterations of the genetic/epigenetic landscape. While prolyl hydroxylases involved in collagen maturation are inhibited, PHD2 that fosters proteasomal degradation of Hif-1α is activated. Consequently, D-2HG is capable of inducing and promoting tumorigenesis and tumor progression. For further details see reviews [18–20]. Mitochondrial IDH3 converts isocitrate to αKG in a NADH-dependent manner. Mutations in the IDH3 isoform have not been reported yet.
D-2HG has been shown to be highly elevated (up to 100-fold) not only in the IDH-mutant tumor cells but also in the tumor microenvironment [21] as well as serum [22] and cerebrospinal fluid [23]. The possibility of D-2HG detection in those biofluids (using mass spectrometry)[23–25] as well as in the tumor tissue (using MRI)[26] provides a valuable tool for non-invasive diagnostics and proved to be more robust in terms of prognostic value. Surprisingly, studies that aimed to elucidate paracrine effects of D-2HG on the tumor microenvironment and particularly the innate and adaptive immune system are less abundant. As tumor cells are capable of shaping their microenvironment to their favor through numerous escape mechanisms, the elucidation of putative tumor-promoting phenomena evoked by exogenous D-2HG is of utmost importance.
Previously, indirect effects of D-2HG produced by IDH-mutated tumors on anti-tumor immunity via their microenvironment have been described. This includes (1) an IKK-independent activation of NFκB signaling in bone marrow-derived mesenchymal stroma cells resulting in the formation of a favorable niche for the D-2HG-secreting AML blasts [27], and (2) the reduction of tumor-infiltrating CD8+ T-cells by the D-2HG-dependent suppression of STAT1 in IDH-mutant glioma cells leading to reduced secretion of T-cell attracting chemokines (CXCL9/10)[28]. Interestingly, the S-enantiomer of 2HG was demonstrated to be produced independent of IDH mutations in T-cells upon T-cell receptor (TCR) triggering in a Hif-1α dependent manner under physiological, hypoxic conditions. Although this increase of intracellular S-2HG to millimolar levels resulted in Hif-1α stabilization, these T-cells showed a restrained cell expansion as well as decreased effector cytokine production and cytotoxicity independent of Hif-1α in vitro. Intriguingly, pre-treatment of adoptively transferred CD8+ T-cells with S-2HG increased their homeostatic proliferation and tumor-controlling capacity. These data indicate a beneficial effect of S-2HG ex vivo (in contrast to the more abundant D-2HG at the tumor site) by maintaining the T-cells’ capacity to proliferate and survive when transferred in vivo which would be otherwise decreased by effector differentiation [29].
The concept of cellular metabolism as a determinant for the phenotype, differentiation, and function of a cell has been shown and established throughout the past years. In T-cells both developmental status and differentiation program are characterized by distinct metabolic profiles optimized for their energetic demands under the given circumstances (Figure 2A). While the cellular metabolism of naïve as well as memory T-cells is characterized by a modest, mixed-fuel oxidative phosphorylation (OXPHOS) for ATP production, activated effector T-cells undergo a glycolytic shift increasing their aerobic glycolysis activity (Warburg effect) while maintaining OXPHOS. Although aerobic glycolysis is less efficient in terms of ATP production per mole glucose it is faster and provides the necessary components for the biosynthesis of other intermediates (such as lipids, nucleic acids and proteins) needed in highly proliferating cells [30]. This increased utilization of glucose together with enhanced secretion of lactate is accompanied by an induction of glutaminolysis as well [31]. The changes in the metabolic phenotype are majorly governed by the enhanced/diminished expression of regulating, nutrient-sensing enzymes such as AMPK, mTOR, and HIF-1α. Distinct from naïve T-cells, memory T-cells metabolism is capable of a rapid recall of aerobic glycolysis after re-challenge [30]. While T-helper effector subsets (Th1, Th2, and Th17) are all favored by a glucose-rich environment due to reliance on aerobic glycolysis, regulatory T-cells are less dependent on glucose and are primarily characterized by fatty acid oxidation and OXPHOS [31].
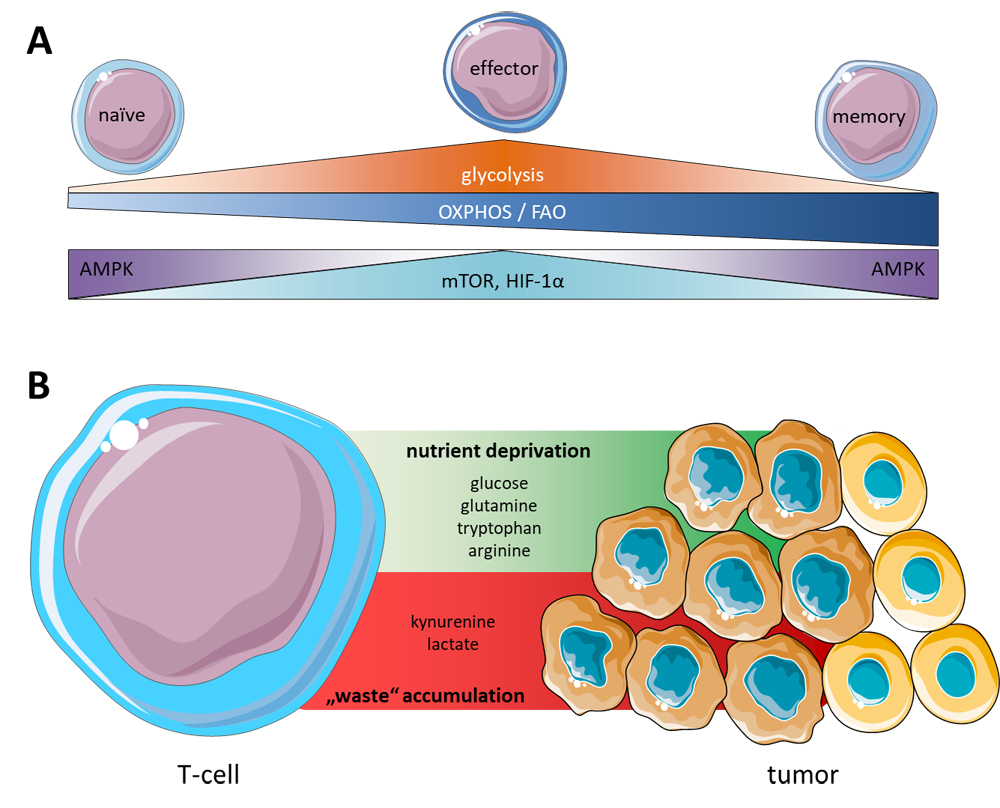 Figure 2. T-cell metabolic profiles and tumor microenvironment. (A) During development and differentiation T-cells rely on different metabolic profiles accompanied by differential expression of governing enzymes AMPK, mTOR, and HIF 1α to meet their energetic demands based on the particular circumstances. (B) The tumor microenvironment is a hostile milieu for effector T-cells shortening nutrient availability and accumulating inhibitory waste products, and thereby creating a tumor-favoring environment.
Figure 2. T-cell metabolic profiles and tumor microenvironment. (A) During development and differentiation T-cells rely on different metabolic profiles accompanied by differential expression of governing enzymes AMPK, mTOR, and HIF 1α to meet their energetic demands based on the particular circumstances. (B) The tumor microenvironment is a hostile milieu for effector T-cells shortening nutrient availability and accumulating inhibitory waste products, and thereby creating a tumor-favoring environment.
The sensitivity of T-cell metabolic programs to intrinsic and extrinsic factors for their functional responses have been shown to be hijacked by a variety of tumors (Figure 2B) including AML and glioma [32,33]. One immune-escape mechanism exploited by tumor cells is the alteration of nutrient availability in the tumor microenvironment. The depletion of nutrients (like glucose and glutamine) or essential amino acids (like tryptophan and arginine) together with the accumulation of “waste” products (like kynurenine and lactate) have all been shown to lead to inhibition of T-cell effector functions, anergy, and cell death [32]. Although many immune-escape mechanisms of tumor cells via a metabolic barrier and/or competition have been illustrated, the entirety of them certainly remains elusive. This is of special interest for immunotherapeutic approaches, which gain more and more importance as putative curative treatments of otherwise incurable cancers. Despite intensive research on immune-cell based therapies there are still a lot of undetermined obstacles dampening the success. Thus, it is of special interest to determine the consequences of oncometabolites on the (metabolic) phenotype and function of T-cells.
Recently, three publications investigating direct effects of exogenous, tumor-derived D-2HG in the context of AML and glioma on T-cells [34–36] have been issued almost simultaneously.
In fact, we and others could show that T-cells are capable of taking up exogenously supplied membrane-impermeable D-2HG [34,35]. So far, studies determining the effects of extracellular D-2HG on different cell types (such as cancer cell lines or cells in the tumor microenvironment) often used cell-permeable, esterified variants like dimethyl-2HG or octyl-2HG. While this enables the analysis of effects evoked by D-2HG on cells that do not produce it themselves, it does not resemble the in vivo situation as D-2HG in its physiological extracellular form comes as a per se cell-impermeable metabolite. Additionally, we showed that exclusively isolated T-cells from IDH-mutated AML patients (that harbor the mutation in the malignant blasts) showed massively increased intracellular amounts of 2HG highlighting the clinical relevance. The capability of D-2HG uptake by T-cells (and other cell types such as murine B-cells, splenocytes and dendritic cells) was further extended by the research of Bunse et al. [35] showing a similar uptake capacity by both non-activated and activated human T-cells. Furthermore, not only the supplemented exogenous D-2HG was taken up by T-cells but also tumor-derived D-2HG secreted by IDH1-mutated glioma cells as shown in a co-culture approach. They demonstrate that this uptake is mediated by the sodium-dependent dicarboxylate transporter 3 (SLC13A3) as sodium-reduction, receptor inhibition or receptor knockout resulted in greatly diminished intracellular D-2HG levels.
Concordant, both studies determined the non-toxicity of exogenous D-2HG up to 20 mM as viability of T-cells did not change upon exposure to the oncometabolite. Although we found no evidence for impaired T-cell proliferation by FACS-based dye dilution or radio-labelled thymidine incorporation, both Bunse et al. [35] and Zhang et al. [36] did so. These differences could be explained by the fact that (1) we analyzed proliferation of T-cells activated by anti-CD2/CD3/CD28 simultaneously with D-2HG treatment, (2) Bunse et al. [35] pre-treated the T-cells with D-2HG prior to stimulation, and (3) Zhang et al. [36] worked with D-2HG concentrations up to 30 mM on murine T-cells that might show a different response and effects on cell viability might not be completely neglectable.
T-cell activation in presence of D-2HG was shown to be reduced (represented by diminished TCR downstream signaling and surface expression of activation marker) in an antigen-unspecific and antigen-specific manner. An impairment of effector cytokine expression (IFN-γ, TNF-α, and Perforin) was not detectable with our antigen-unspecific simultaneous stimulation/D-2HG treatment regimen. Intriguingly, INFγ secretion after antigen-specific stimulation of both human and murine T-cells was indeed affected by D-2HG as shown by Bunse et al. They also found reduced IFNγ production of murine T-cells co-cultured with IDH1-mutated astrocytes after stimulation with PMA/Ionomycin indicating that tumor-derived D-2HG impairs effector cytokine production. However, other direct or indirect T-cell inhibiting effects by the IDH1-mutated astrocytes apart from the secreted oncometabolite cannot be completely excluded. Still, Zhang et al. [36] also showed a concentration-dependent inhibition of IFNγ secretion of murine T-cells by exogenous D-2HG. They could also demonstrate that the previously published reduction of T-cell infiltration into the tumor site was not solely mediated by reduced chemotaxis due to D-2HG-provoked chemokine suppression in the tumor cells. Instead, they saw a direct effect of D-2HG on T-cell migration in an inhibitory manner but the underlying mechanisms remain elusive.
The ability of D-2HG to impair TCR signaling and downstream effects as well as the fact that D-2HG is structurally similar to αKG raises the question if the obtained results are mediated on an immunometabolic level.
We found a dose-dependent, reversible induction of glucose utilization, which was accompanied by a reduced secretion of lactate indicating an increased flux of glucose to the TCA cycle instead of aerobic glycolysis. In a metabolomic profiling Bunse et al. also found elevated levels of hexoses in D-2HG treated CD4+ and CD8+ T-cells irrespective of the activation status. Indeed, intracellular amounts of TCA cycle intermediates were found to be increased after D-2HG treatment in both studies. It can be speculated here that this may be caused by the physiological function of D-2HG dehydrogenase (D2HGDH) converting D-2HG to αKG leading to anaplerosis of the TCA cycle. Moreover, glutaminolysis as another anaplerotic switch for the TCA cycle was shown to be enhanced as exemplified by the increased expression of the rate-limiting enzyme glutaminase (GLS)[34] as well as elevated levels of intracellular l-glutamine [35]. On the other hand, expression of PDK1 (that blocks the entry of pyruvate into the TCA cycle) and LDHA (that converts pyruvate to lactate) was significantly reduced by D-2HG [34]. Accordingly, we found an increase of mitochondrial respiration and a decrease of aerobic glycolysis by D-2HG as measured by real-time metabolic flux analysis. Nevertheless, D-2HG did not provoke a glucose-dependency in the T-cells as diminished glucose availability as well as inhibition of the glycolysis rate-limiting enzyme hexokinase had similar effects on viability and proliferation in both untreated and D-2HG treated T-cells. On the contrary, Bunse et al. found a reduction of oxygen consumption by D-2HG pre-treated T-cells upon activation [35] indicating a high flexibility of T-cell metabolic responses towards the oncometabolite.
Mammalian target of rapamycin (mTOR) holds a key role in cellular metabolism regulating mitochondrial oxygen consumption [37], glucose uptake [38], and nutrient utilization [39] by dictating the expression and or activity of metabolic enzymes [40]. In fact, enhanced mTOR signaling was found in mutant IDH-expressing cell lines mediated by the inhibition of the αKG-dependent enzyme KDM4A [41]. In line with the observed metabolic phenotype, T-cells stimulated in the presence of D-2HG also showed enhanced mTOR signaling (illustrated by the phosphorylation of both mTOR and its down-stream target 4EBP1) despite unaltered MTOR gene expression. Actually, a negative impact of the oncometabolite on the protein expression of the endogenous mTOR inhibitor DEPTOR (via the described competitive inhibition of αKG-dependent KDM4A) was found to be the responsible driver [34,41]. In fact, D-2HG induced a mTOR-dependency as rapamycin renders T-cells vulnerable to D-2HG treatment in terms of glucose uptake and proliferation. Another metabolic master regulator and sensor of AMP/ADP:ATP ratio, AMPK [42], was found to be phosphorylated and thus activated in D-2HG treated T-cells due to a lack of total intracellular ATP [35]. Increased AMPK signaling has been shown to be amongst others responsible for catabolic processes such as enhanced glucose uptake and utilization, fatty acid uptake and oxidation, mitochondrial biogenesis and enhanced mTOR signaling [43]. Additionally, AMPK was shown to inhibit ornithine decarboxylase 1 (ODC1) activity, the rate-limiting enzyme of polyamine biosynthesis. As a consequence, intracellular levels of ornithine accumulated while its decarboxylation products putrescine and spermidine were reduced. Interestingly, those products were proven to have potent immuno-stimulatory effects [44] and their lack can at least partly explain the observed defects in T-cell proliferation and activation [45].
The transcriptional activity of HIF-1α is also known to be regulated by mTOR [46]. However, D-2HG has been shown to destabilize Hif-1α protein by the induction of αKG-dependent prolyl hydroxylases leading to its proteasomal degradation [10]. Accordingly, we found a decrease of intracellular Hif-1α protein caused by D-2HG treatment in activated T-cells despite enhanced mTOR activity and unaltered HIF1A gene expression. Consequently, Hif-1α target genes including the aforementioned PDK1 and LDHA were lower expressed.
Taken together, D-2HG is capable of shaping cellular T-cell metabolism on different levels influencing central regulators such as mTOR, AMPK, and Hif-1α, which can have profound consequences for their functional properties.
As mentioned earlier, both the developmental stage and the differentiation of T-cells rely on different metabolic profiles. Thus, it is tempting to speculate that D-2HG mediated alterations in T-cell metabolism can lead to different compositions of T-cell subsets. The shift of T-cell metabolism from glycolytic to more oxidative by D-2HG treatment during activation might favor memory formation and/or regulatory T-cells (Tregs). As a matter of fact, we and Zhang et al. [36] found increased frequencies of CD4+ CD25+ FoxP3+ Tregs after in vitro stimulation of human and murine T-cells in the presence of D-2HG, respectively. However, it remains to be determined if Tregs are induced by the D-2HG mediated metabolic switch or if they inherit a yet unknown survival benefit (e.g., similar to ROS [47] or lactate [48]) as concentrations beyond 20 mM (which we found to be toxic for naïve T-cells) increased Treg frequencies even stronger (data not published). Interestingly, an earlier study from 2017 described a shift in the balance of Th17 and Tregs in favor of Th17 cells after intracellular accumulation of or external treatment with D-2HG which finally lead to epigenetic alterations at the FoxP3 promoter [49]. Notably, D-2HG accumulation did not result from IDH-mutations but from transamination. Resulting intracellular levels of D-2HG were determined to be approx. 0.2 mM which is about 100-fold less than in the malignant situation of IDH-mutated tumors. Moreover, T-cells in those experiments were cultured under Th17- or Treg-inducing conditions and treated with a cell-permeable variant of D-2HG. Thus, these studies by Xu et al. [49] represent a separate situation irrespective of D-2HG being an oncometabolite in the tumor context and as such cannot be directly compared to the other discussed findings. Apart from the Treg-induction, alterations of the effector/memory compositions in the in vitro cultures by D-2HG could not be determined. However, Bunse et al. found increased CD4+ naïve and reduced CD4+ memory T-cell transcripts in a differential expression profiling of IDH wt and IDH mutant gliomas [35]. Yet, other factors apart from tumor-secreted D-2HG cannot be completely excluded as causatives for the differential expression.
Beyond that, Hif-1α has already been linked to the formation of Th17 cells [50]. Consequently, the already mentioned reduction of Hif-1α protein by D-2HG resulted in a diminished expression of target genes such as RORC and IL17A as well as in a reduction of IL-17 secretion. Accordingly, frequencies of Th17 cells were also reduced by D-2HG treatment in vitro. Ultimately, in the context of AML Th17 frequencies were significantly reduced ex vivo in samples derived from patients carrying an IDH2 mutation as compared to non-mutated AML cases [34]. However, the role of Th17 cells for prognosis and progression of AML is still under debate. Though, their essential role to fight infections during AML progression, which are a major cause of morbidity and mortality, certainly highlights a clinical relevance.
In the context of the glioma, the reduced levels of total intracellular ATP mediated by exogenous D-2HG not only affected AMPK but also reduced ATP-dependent T-cell receptor (TCR) down-stream signaling such as Fyn, JNK, and PLCγ1. This in turn reduced the Ca2+ influx and inhibited translocation of NFAT and NFκB to the nucleus [35] which explains the observed reduction of T-cell activation and effector cytokine production, e.g., IL-2, IL-4, IL-10, IL-22, IL-31, Granzyme B, IFNγ, and TNFα, as those are known target genes of the two transcription factors.
Those findings were further shown to be relevant in vivo in mutant IDH glioma and sarcoma models by Bunse et al. [35]. They found reduced T-cell infiltration with diminished effector cytokine production and NFAT signaling exclusively in T-cells of mutant IDH tumors in humanized mouse models. Furthermore, specific vaccination with a peptide of the mutant IDH resulted in an increased rejection rate. Interestingly, peripheral lymphocyte responses of vaccinated mice did not differ between the wildtype and mutant IDH setup indicating a restriction of D-2HG mediated T-cell impairment to the site of the tumor. In fact, in glioma and glioblastoma D-2HG levels have been reported to be locally elevated up to 35 mM [21]. The relevance of tumor-derived D-2HG on anti-tumor T-cell response was further proven by application of two different mutant IDH inhibitors, i.e., BAY1436032 and AG-5198, as those elevated intratumoral T-cell proliferation and alleviated inhibition of tumor growth. Ultimately, overall survival of mutant IDH glioma-bearing mice was improved solely by the simultaneous administration of a mutant IDH inhibitor together with PD-1 inhibition [35] highlighting new potential therapeutic regimens for the treatment of patients.
Importantly, all observed effects were shown to be specific for the d-enantiomer of 2HG [34,35] and not evoked by epigenetic alterations in the T-cells [35].
Taken together, the results of the three recent studies emphasize the importance of D-2HG not only as an autocrine but also paracrine oncometabolite capable of shaping the tumor microenvironment in several facets adding to the common concept of tumor immune escape mechanisms (Figure 3). However, it becomes also clear that D-2HG certainly has entity- and niche-specific effects, which also depend on the developmental and activation status of the target T-cells. Interestingly, this can potentially add to the conflictive prognostic impact of IDH mutations in AML and glioma. In fact, mutant IDH-expressing glioma patients have a better prognosis compared to wild-type counterparts of the same histological grade [51]. This might seem contradictory to the presented findings of immunometabolic effects by D-2HG on T-cells and T-cell infiltration at the tumor site. However, it is speculated that IDH-wildtype gliomas might harbor additional mutations and alterations leading to the worse prognosis [28] which certainly needs more experimental evidence. Moreover, there are indications that IDH-mutated malignant cells are surveyed by immune cells but locally elevated D-2HG concentrations at the tumor site break this immune surveillance [35]. Nevertheless, those recent findings will have a great impact especially in view of targeted therapies against mutant IDH in combination with immune-based approaches as shown by the improved outcomes using mutant IDH inhibitors together with PD-1 blockade in pre-clinical approaches. Additionally, the reduction of pro-inflammatory Th17 cells accompanied by an increased Treg frequency in the context of AML could highlight the importance of that oncometabolite for anti-tumor immunity as well as infections that are a major cause of mortality and morbidity during disease progression. Finally, current and upcoming clinical trials using inhibitors of mutant IDH as well as mutant IDH-based peptide vaccinations should be broadened to determine not only the tumor-intrinsic effect of the inhibitor but also a potential therapeutic benefit in restoring anti-tumor immune responses. Future studies will certainly be able to elucidate if and how inhibition of mutant IDH can enhance the efficacy of current and ongoing immunotherapeutic approaches.
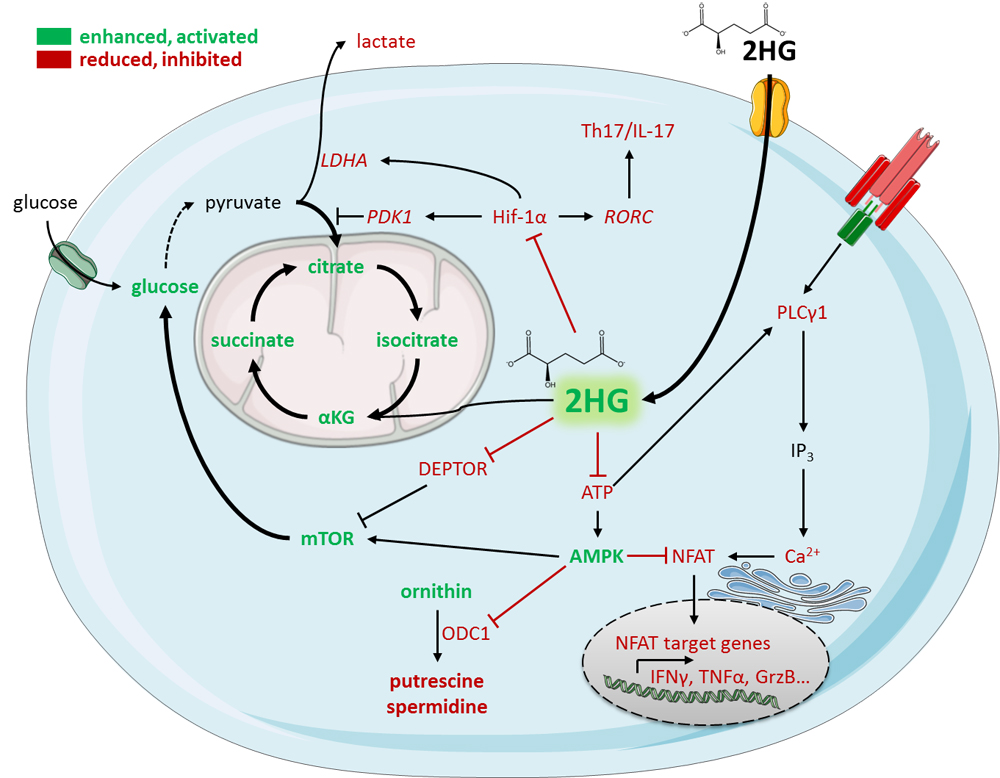 Figure 3. D-2HG mediated metabolic and functional changes in T-cells. T-cells are capable to take up
D-2HG via solute-carrier proteins. Intracellular accumulation of D-2HG leads to Hif-1α destabilization resulting in an increased influx of pyruvate to the TCA cycle and a diminished Th17 formation. The TCA cycle is further fueled by enhanced glucose utilization due to mTOR signaling activation and anaplerotic replenishment of αKG. At the same time, the total ATP pool in the cell is reduced leading to AMPK activation and dysfunctional ATP-dependent TCR downstream signaling. Both effects result in a prohibited calcium-dependent NFAT signaling leading to an impairment of target gene expression. Additionally, AMPK inhibits polyamine biosynthesis leading to proliferation defects. All effects taken together lead to an impaired anti-tumor immunity.
Figure 3. D-2HG mediated metabolic and functional changes in T-cells. T-cells are capable to take up
D-2HG via solute-carrier proteins. Intracellular accumulation of D-2HG leads to Hif-1α destabilization resulting in an increased influx of pyruvate to the TCA cycle and a diminished Th17 formation. The TCA cycle is further fueled by enhanced glucose utilization due to mTOR signaling activation and anaplerotic replenishment of αKG. At the same time, the total ATP pool in the cell is reduced leading to AMPK activation and dysfunctional ATP-dependent TCR downstream signaling. Both effects result in a prohibited calcium-dependent NFAT signaling leading to an impairment of target gene expression. Additionally, AMPK inhibits polyamine biosynthesis leading to proliferation defects. All effects taken together lead to an impaired anti-tumor immunity.
MB and DM wrote the manuscript. MB created the figures using Servier Medical Art licensed under a Creative Commons Attribution 3.0 License (https://smart.servier.com/).
The authors declare that there are no conflicts of interest.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
Böttcher M, Mougiakakos D. Immunometabolic Regulation of Anti-Tumor T-Cell Responses by the Oncometabolite D-2-Hydroxyglutarate. Immunometabolism. 2019;1:e190007. https://doi.org/10.20900/immunometab20190007.

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




