Location: Home >> Detail
J Psychiatry Brain Sci. 2019;4:e190007. https://doi.org/10.20900/jpbs.20190007
 ,
Mihran J. Bakalian 1,
Andrew J. Dwork 1,2,
Eli Min 1,
J. John Mann 1,2,
Victoria Arango 1,2
,
Mihran J. Bakalian 1,
Andrew J. Dwork 1,2,
Eli Min 1,
J. John Mann 1,2,
Victoria Arango 1,2
1 Department of Molecular Imaging and Neuropathology, New York State Psychiatric Institute, New York, NY 10032, USA
2 Department of Psychiatry, Columbia College of Physicians and Surgeons, New York, NY 10032, USA
* Correspondence: Mark D. Underwood, Tel.: +1-646-774-7545.
Alcohol increases inhibitory neurotransmission, an effect mediated through GABA receptors. With chronic alcohol exposure, the inhibitory effects diminish. Glutamic acid decarboxylase (GAD) catalyzes glutamate in the synthesis of GABA. We sought to determine the amount of GAD65/67 mRNA in anterior cingulate cortex (BA24) and orbital prefrontal cortex (BA45) of medication-free alcoholics and nonpsychiatric controls postmortem.
Studies were performed in 16 pairs of nonpsychiatric controls and alcoholics, matched for age, sex and PMI. DSM-IV diagnosis of alcohol use disorder (AUD) was made by the SCID I in a psychological autopsy. Frozen blocks of BA24 or BA45 were sectioned (10 µm) for in situ hybridization of 35S-labelled riboprobe for GAD65/67 mRNA and autoradiograms were analyzed by quantitative densitometry. Three isodensity bands of labeling were evident, with different relative amounts of GAD65 and GAD67 (outer and inner, predominantly GAD65, intermediate predominantly GAD67), and the isodensity bands were analyzed separately.
GAD65/67 mRNA levels were not different between alcoholics and controls in the gray matter of BA24 (p = 0.53) or BA45 (p = 0.84) or in any of the three isodensity bands in which the GAD65/67 mRNA was distributed. GAD65/67 mRNA in white matter underlying either region was also not different in alcoholics (p > 0.05). GAD65/67 mRNA levels did not correlate with age, sex or duration of alcoholism in either BA24 or BA45.
Effects on inhibitory neurotransmission in alcoholics do not appear to be associated with change in the levels of GAD65 or GAD67 mRNA.
Alcohol consumption produces dose-dependent inhibition of the central nervous system (CNS) [1]. Alcohol has complex effects on several organs, in particular, the brain. Alcohol has effects on lipid bilayers and affects multiple neurotransmitters in multiple brain regions (see [2,3] for review).
The CNS inhibition brought about by alcohol exposure is attributed to inhibition of excitatory glutamatergic neurotransmission [4,5] and also by ethanol acting as an indirect gamma-aminobutyric acid (GABA) agonist [6]. GABA is the main inhibitory neurotransmitter in the brain, and ethanol acts through actions on GABAA receptors (e.g., [7,8], decreasing neuronal excitability (see [9] for review). With chronic alcohol exposure, there is neuronal adaptation and the inhibitory effects are reduced [5].
It has been hypothesized that ethanol acts on several neurotransmitters in a combination that is reinforcing by activating reward centers and also by reducing stress [10]. The prefrontal cerebral cortex (PFC) is a critical brain region in the reinforcing effects of alcohol [11,12] and that predisposition to the addictive effects of alcohol exposure may arise from reduced behavioral-cognitive inhibition [13,14].
The PFC functions to provide executive function through behavioral inhibition mediated through GABAergic inhibitory interneurons, and in alcoholics there is reduced behavioral inhibition [15,16]. Glutamic acid decarboxylase (GAD) is the biosynthetic enzyme of GABA and exists in two major isoforms (GAD65 and GAD67), encoded by two different genes (GAD2 and GAD1) with different relative anatomical distributions in cerebral cortex [17]. Outer and inner cortical layers are predominantly GAD65 with a greater prevalence in the neuropil and intermediate layers are predominantly neuronal soma with relatively more GAD67 [17]. GAD decarboxylates glutamate resulting in the synthesis of GABA and CO2 [18–20]. We sought to determine the amount of GAD65/67 mRNA in the postmortem prefrontal cortex as an index of inhibitory GABAergic effects in alcoholics.
The brains used in the study, as well as the diagnoses and other clinical demographic information, were provided by the Molecular Imaging and Neuropathology Division at the New York State Psychiatric Institute. The brain collection, psychological autopsies, and the use of the brain tissue was approved by the Institutional Review Board of the New York State Psychiatric Institute or the University of Pittsburgh. Tissue Collection for Neuropathologic Studies of Major Psychiatric Illness, NYSPI Protocol #7351, was last approved 8/29/2018. First approved 7/18/2001 as Protocol #4127. The Institutional Review Board (IRB) of the New York State Psychiatric Institute made determinations (in project identification codes #5073R (5/9/2008) and #5154R (8/16/2011)), that the experiments performed were not human subjects research.
Experiments were performed in 16 pairs of sudden-death, nonpsychiatric controls (C) or alcohol use disorder cases (AUD) matched for age (C: 40 ± 4 years; AUD: 40 ± 4; mean ± SEM, p > 0.05) and sex (26 males and 6 females). Neither postmortem interval (PMI) (C: 15.0 ± 1.2 h; AUD: 13.5 ± 1.3; t = 0.834, df = 30, p = 0.411) nor brain pH (C: 6.582 ± 0.066; AUD: 6.655 ± 0.055; t = 0.856, df = 30, p = 0.4) was significantly different between groups. Both the controls and AUD cases were diagnosed retrospectively by psychological autopsy using SCID I [21]. Final DSM-IV diagnosis of AUD was made through consensus of experienced clinicians reviewing SCID I, administered in the third person to next-of-kin and other relatives, and all other available data including medical records. Toxicology was provided by the Medical Examiner and was negative in the controls (except for 2 cases with cannabis); alcohol was present in some of the AUD cases and one alcoholic was positive for benzodiazepines (see Table 1). Cases with positive neuropathology were excluded.
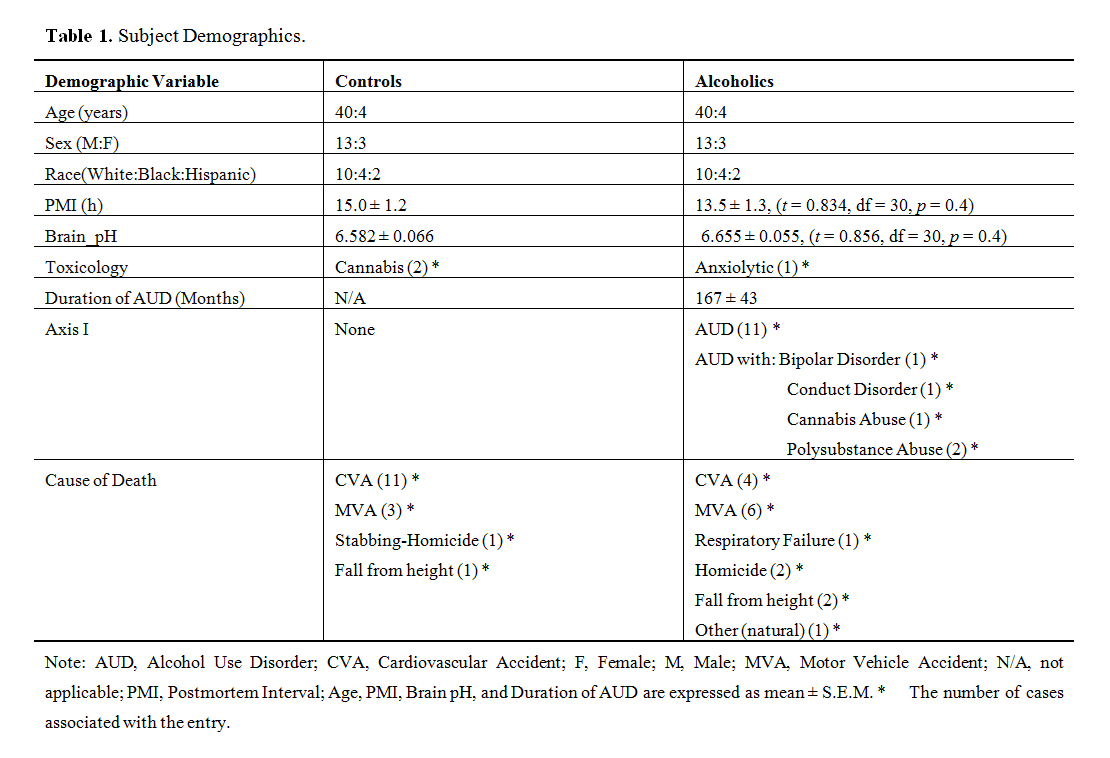 Table 1. Subject Demographics.
Table 1. Subject Demographics.
Brains were collected at autopsy. Following removal from the calvarium, the brainstem was removed and the forebrain was bisected in the mid-sagittal plane; the hemispheres were hand cut into several slabs approximately 2 cm thick. The right hemisphere was frozen while the left hemisphere was fixed in formalin. Frozen slabs containing the prefrontal cortex from the right hemisphere were used in the present study. Slabs were stored frozen (−80 °C) until selected for the study. Slabs were sectioned in a cryostat and sectioned at 10 µm. Anterior cingulate cortex (BA24) and orbital prefrontal cortex (BA45/47) cryosections were mounted on gelatinized glass slides (2.5 cm × 7.5 cm), and the slides were stored at −80 °C until use.
In situ hybridization. Sections were fixed in cold 4% paraformaldehyde, washed in DEPC-treated phosphate buffered saline (PBS), and treated with triethanolamine buffer containing 0.25% acetic anhydride and rinsed 2 times in saline-sodium citrate buffer. Slides were dehydrated in ethyl alcohols 50%, 70%, 95% and 100%, delipidated in chloroform, and rehydrated. 35S-radiolabeled antisense riboprobe specific for GAD65/67 mRNA, mixed with hybridization buffer, was applied to each section (100 µL per section, containing 2 × 106 cpm). Hybridization was performed at 55 °C for 18–20 h in humidified chambers. The sections subsequently went through stringent washes in different concentrations of SSC buffer, formamide buffer at 55 °C and treated with RNase A at 37 °C. The sections then were exposed to autoradiographic film (Kodak, Biomax MR, Rochester, NY, USA) in cassettes for 29 to 30 days. Subsequently, slides were dipped in Kodak nuclear track emulsion (1:1, emulsion: H2O) and exposed for 2 weeks at 4 °C in sealed boxes with desiccant. The emulsion-coated slides were developed with Kodak D-19 developer and stained in Hematoxylin & Eosin.
Autoradiograms were analyzed for labeled mRNA in BA24 and BA45/47 in gray and white matter by quantitative densitometry (MCID, Imaging Research, St. Catharines, Canada). Optical density was calibrated to µCi of radioactivity/gram by co-exposure of the autoradiographic films with radioactive standard slides. The resulting relationship between optical density and concentration of radioactivity was linear through the calibrated range. A digital threshold was applied to delineate gray matter from white matter, and Brodmann area was user-defined with reference to adjacent Nissl-stained sections. Three isodensity bands of GAD65/67 distribution were apparent and were user-defined and sampled separately.
Comparisons between AUD cases and controls were made using linear models with age as a covariate. Paired t-tests were also used since cases and controls were matched for age, sex and PMI. Measurements were made in 2 brain regions (BA24 and BA45), each with 3 isodensity bands (outer, intermediate, inner) and white matter underlying each region, so correction for multiple comparisons was performed using a Bonferroni correction and setting the p-value for statistical significance at p = 0.01. Relationships between the amount of mRNA and continuous variables (age, duration of alcoholism, PMI) were examined using Pearson correlation coefficients. We performed power analyses using the G*Power software version 3.1.9.4 [22]. Data are presented as mean ± standard error of the mean (S.E.M.).
GAD65/67 mRNA expression was evident throughout the gray matter of BA24 and BA45 in both controls and AUD cases (Figure 1). At higher magnification, the silver grains in the photographic emulsion were located largely over selected neurons, and silver grains in the neuropil were present in a lower density than over the soma of labeled neurons (Figure 2). BA24 and BA45 gray matter have similar amounts of GAD65/67 (Figure 3). GAD65/67 was present at a much lower concentration in the underlying white matter, with BA45 white matter having significantly more GAD65/67 than BA24 white matter (t = 4.404, df = 34.582, p < 0.001, Figure 3).
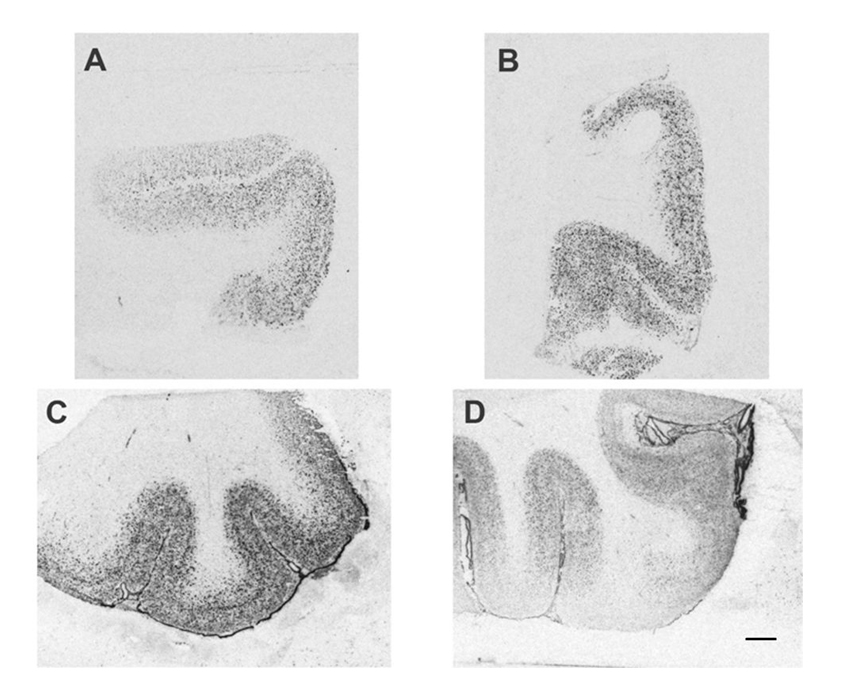 Figure 1. GAD65/67 mRNA expression in BA24 (A, B) and BA45/47 (C, D) in a representative control (A, C) and alcoholic (B, D) visualized by in situ hybridization histochemistry. Note the labeled GAD65/67 mRNA is localized throughout the gray matter and is greater in BA45 than in BA24. Scale bar = 2 mm.
Figure 1. GAD65/67 mRNA expression in BA24 (A, B) and BA45/47 (C, D) in a representative control (A, C) and alcoholic (B, D) visualized by in situ hybridization histochemistry. Note the labeled GAD65/67 mRNA is localized throughout the gray matter and is greater in BA45 than in BA24. Scale bar = 2 mm.
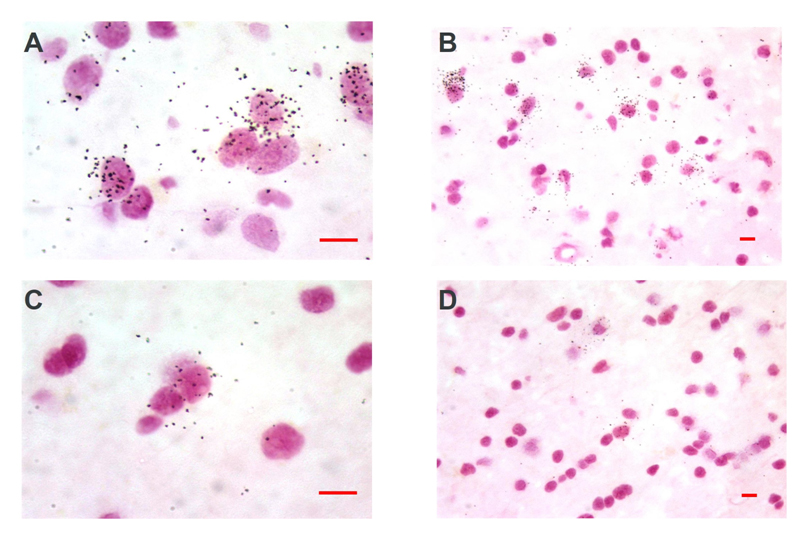 Figure 2. Photomicrographs of tissue section hybridized with [35S]-labeled ribonucleotide probe and developed for silver grains in a representative control (A, B) and alcoholic (C, D). Note the silver grains over cells.
Figure 2. Photomicrographs of tissue section hybridized with [35S]-labeled ribonucleotide probe and developed for silver grains in a representative control (A, B) and alcoholic (C, D). Note the silver grains over cells.
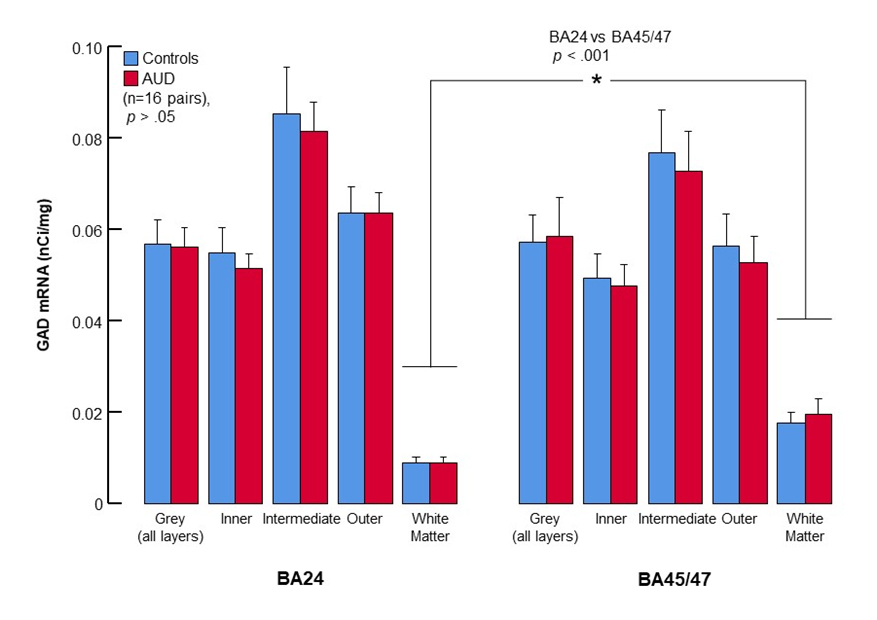 Figure 3. Bar plots of GAD65/67 density in Brodmann Areas 24 and 45/47. Data are presented as mean ± S.E.M. Note the lack of distinction between controls and AUD cases regardless of region sampled.
Figure 3. Bar plots of GAD65/67 density in Brodmann Areas 24 and 45/47. Data are presented as mean ± S.E.M. Note the lack of distinction between controls and AUD cases regardless of region sampled.
AUD cases have a comparable amount of GAD65/67 mRNA as controls in the gray matter of BA24 (F1,14= 0.421, p = 0.53) and BA45 (F1,13 = 0.045, p = 0.84). In BA24, the effect size of the difference between the AUD and controls was 0.17 with an achieved statistical power of 0.382 to detect a difference at α = 0.05 (paired t-test). In BA45/47, the effect size of the difference was 0.21 with 54% achieved power (paired t-test). In white matter underlying either Brodmann area there was also no significant difference between controls and AUD cases (t = 0.003, df = 24 (BA24) and t = 0.484, df = 26 (BA45)). Controls and AUD cases also did not significantly differ with inclusion or exclusion of the 2 cases positive for cannabis or the 1 with benzodiazepines (data not shown).
GAD65/67 mRNA levels did not change with age in either BA24 or BA45 (r = −0.28, p = 0.13; r = −0.28, p = 0.16, respectively) in all cases combined or in control or alcoholic groups separately. GAD65/67 mRNA did not change with duration of alcoholism in BA24 (r = 0.234, p = 0.515) or in BA45 (r = 0.468, p = 0.203). GAD65/67 mRNA levels did not correlate with the alcohol abuse rating in BA45 (r = 0.017, p = 0.945) or BA24 (r = −0.244, p = 0.287).
GAD65/67 mRNA did not correlate with PMI or brain pH in BA45 or BA24 or in the underlying white matter (p > 0.05, all measures and regions). Males had a similar density of GAD65/67 mRNA as females in BA24 (males: 0.057 µCi/g ± 0.004; females: 0.053 µCi/g ± 0.002; t = 0.983, df = 29.956, p = 0.334) and in BA45 (males: 0.057 µCi/g ± 0.007; females: 0.062 µCi/g ± 0.004; t = 0.457, df = 26, p = 0.651).
In the present study, we did not detect a statistically significant difference in levels of GAD65/67 mRNA in orbital PFC (BA45/47) or in anterior cingulate cortex (BA24) in AUD cases compared with non-alcoholic psychiatrically normal controls.
GABAergic neurotransmission contributes to the effects of ethanol by potentiating neuronal inhibition through actions on GABAA type receptors (see [23] for review). With chronic ethanol exposure, brain levels of GABA are decreased [24,25] and the concentration of GABA receptors is increased [26,27], specifically of the GABAAα1 and GABBAAβ3 subtype [28,29]. The similar levels of GAD mRNA we found in alcoholics suggests that the lower levels of GABA with chronic ethanol exposure is not due to reduced GAD expression.
Lower GAD67 mRNA is reported in schizophrenia and bipolar disorder [30–32] as well as in major depression [33]. However, the presence of comorbid psychiatric illness in our sample of AUD as an explanation of our findings is not likely because our cases were evaluated by a full SCID I and SCID II in the course of a psychological autopsy using our validated method [21] and any cases or controls with major comorbid diagnoses were excluded.
We found that GAD65/67 mRNA level is not related to the duration of alcoholism and we found no change in GAD65/67 mRNA with age the alcoholics. Others have reported a decrease in GAD mRNA with age in hippocampus [34] and dorsolateral prefrontal cortex [35]; and no relationship between GAD mRNA and age in anterior cingulate cortex [32] or BA9 [36] in human brain postmortem. Therefore, age does not appear to be an important factor in the estimation of GAD65/67 mRNA in controls or with the duration of alcohol abuse, at least not in the age range we used or in the orbital PFC or anterior cingulate cortex.
Sex differences in GAD are reported in rat neonates and GABA contributes to the development of sexually dimorphic anatomy and behaviors [37], however, the sexual differences are gone by postnatal day 15. Heckers and colleagues [34] found that males had a higher density of GAD mRNA positive neurons in the hippocampal CA1 region and more GAD mRNA expression in neurons of the CA4 region. Hashimoto et al. [38] found no differences in GAD67 mRNA between males and females in BA 9 in their study of schizophrenics, while Duncan et al. [35] found more GAD67 mRNA in males in dorsolateral prefrontal cortex (BA region was not specified). Interestingly, alcohol dependence is more prevalent in males than females [39]. Regardless, the sex-dependent difference in alcohol dependence does not appear to be due to GAD mRNA.
GAD65 and GAD67 have different intracellular labeling patterns with GAD65 having a predominance in the neuropil and GAD67 in the neuron soma [17]. We chose a riboprobe for both GAD isoforms for practical reasons in that the labeling in our postmortem tissue was superior to either isoform alone. As a result, we cannot distinguish whether any possible changes were masked due to offsetting changes in one isoform or the other. We believe this is of limited importance since we were labeling the mRNA isoforms and not the proteins themselves. It is perhaps of greater functional significance that the 2 GAD isoforms are thought to be associated with 2 different pools of GABA, a neurotransmitter pool arising from GAD activity in the nerve terminals and a metabolic pool from GAD67 in the neuron cell body [20]. It is unclear as to the extent that these contributions are absolute, and it is thought that both isoforms can contribute to both pools, but the differences for the purposes of the present study are moot as we did not distinguish between the two isoforms and we did not detect any difference in the levels of GAD mRNA in AUD compared to controls.
We did not measure neuron number or density, raising the possibility that there are increases in GAD mRNA per neuron in the AUD cases if they have had a loss of neurons as has been reported elsewhere in the literature [40–42]. We think this is unlikely in our study population since loss of neurons in the cerebral cortex in alcoholics is typically only seen in severely alcoholic cases such as those with Wernicke’s Encephalopathy or with gross neuropathology or comorbid neurodegenerative disease [43–45]. Such cases should have been detected by the neuropathology screen and psychological autopsy procedure which includes medical history, and would have been excluded from our study.
We cannot rule out the possibility that the alcoholics had lower, or even higher, GAD mRNA levels prior to alcohol exposure, and that the exposure to alcohol changed GAD mRNA levels to those similar to normal controls. The subsequent normal level of GAD mRNA with chronic ethanol exposure could then be a compensatory response to normalize GABA synthesis levels. The compensation, in turn, could be a biological component of tolerance to ethanol and a reduced sensitivity over time to alcohol’s effects. Such possibilities highlight a limitation of postmortem studies, namely not being able to determine cause and effect relationships. For example, “chronic” ethanol administration do not affect GAD mRNA levels in the rat cerebral cortex [46]. However, the “chronic” model in the rat was 14 consecutive days of exposure; such a model in the rat may not capture the molecular changes associated with years or decades of ethanol exposure in humans. Moreover, the cellular organization of the rat orbital cortex may not capture the complexity of the human orbital cortex.
We cannot rule out the possibility that GAD mRNA is altered with AUD in other brain regions. We chose to examine the orbital prefrontal cortex and the anterior cingulate cortex because of their role in behavioral inhibition and emotion regulation. It is possible however, that GAD and GABA are involved in AUD, but in other regions that we did not study, or in brain reward circuits such as the nucleus accumbens, hippocampus or amygdala [47]. It may be there is a mismatch between the GAD mRNA expression and GAD protein, not only anatomically but also temporally. GAD mRNA may not be different in expression in AUD, but the transcription into protein may be altered. We chose to measure GAD mRNA and not GAD protein because GAD protein is more susceptible to postmortem degradation [36,48]. In addition, mRNA in situ hybridization and autoradiographic sampling of regions of interest is amenable to quantitation of the 35S-labeled probes and a regional analysis. Differences in mRNA expression may have occurred at a different point in time in the illness, and we looked only after adaptation had occurred. These possibilities remain unaddressed and must be considered limitations of the present study.
We did find less GAD mRNA expression in the white matter underlying BA24 and BA45/47 compared with gray matter. This is expected since mRNA is localized to neurons or their process [49,50]. There was no difference between controls and AUD, only between the two regions. We think the difference between white matters is real and not an artifact and the difference is most likely the result of different orientation of white mater tracts, as is seen in differences in fractional anisotropy by diffusion tensor imaging [51]. White matter tracts in cross section will present less mRNA in cell bodies than tracts cut longitudinally.
Lastly, we performed power analysis to determine whether the present study was sufficiently powered to detect a difference between the control and AUD group given the effect size and variability observed and whether the results were negative or merely inconclusive. In BA45/47, the effect size of 0.21 was small (see [52]). In BA24, the effect size of 0.17 was smaller. Given the variability in the measure in each of the brain regions, the difference between AUD and controls is likely not physiologically relevant even if the study sample was to be significantly enlarged and we therefore conclude the findings here are negative. More studies are needed to replicate the present findings in a much larger group of subjects and measurement of GAD protein would better serve to more definitively determine whether and how GAD mRNA expression and GABA are altered in AUD.
VA, JJM and MDU designed the study. All authors contributed to the acquisition, analysis and interpretation of the data and drafted the manuscript, revising it critically for important intellectual content. MDU and MJB wrote the paper with input from all authors. All authors provided final approval of the published version of the manuscript, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
J. John Mann receives royalties from the research foundation for mental hygiene for commercial use of the C-SSRS.
This work was supported by grants from the National Institute of Alcoholism and Alcohol Abuse (AA11293), the National Institute of Mental Health (MH40210, MH62185) and the Diane Goldberg Foundation. Some of the brain samples and their psychiatric characterization and storage were funded by NIH grant MH64168.
We acknowledge Steve Ellis’ support for statistical interpretations and power analyses.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
Underwood MD, Bakalian MJ, Dwork AJ, Min E, Mann JJ, Arango, V. GAD mRNA in Orbital Prefrontal Cortex and Anterior Cingulate Cortex in Alcoholics Compared with Nonpsychiatric Controls: A Negative Postmortem Study. J Psychiatry Brain Sci. 2019;4:e190007. https://doi.org/10.20900/jpbs.20190007

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




