Location:Home >> Detail
J Psychiatry Brain Sci. 2016;1(4):3; https://doi.org/10.20900/jpbs.20160017
 ,
Ruihuan Xu6*
,
Ruihuan Xu6*
1 Division of psychology, Shenzhen Children’s Hospital, Shenzhen, Guangdong, China
2 Division of Shenzhen Children’s Hospital, Shenzhen, Guangdong, China
3 Tianjin Jinnan Xiaozhan Hospital
4 Division of Gastroenterology, Shenzhen Children’s Hospital, Shenzhen, Guangdong, China
5 Shenzhen Following Precision Medical Research Institute, Shenzhen, Guangdong, China
6 Clinical Laboratory, Longgang Central Hospital of Shenzhen, Guangdong, China
7 Shenzhen Imuno Biotech Co.,Ltd
8 Division of Neonatology, Children’s Hospital of Fudan University, Shanghai, China
9 Key Laboratory of Birth Defects, Children’s Hospital of Fudan University, Shanghai, China
*Correspondence: Mingwang Wang; Ruihuan Xu.
Chromosomal microarray (CMA) has been suggested for first tier clinical diagnostic test for ASD, with hundreds of genes being associated with ASD by High-throughput Sequencing (HTS). Whole Exome Sequencing (WES) was used to combine with CMA to identify clinical relevant variants in ASD. In our previous work, we used trio (father, mother and proband) based WGS (Whole Genome Sequencing) to reveal clinical relevant de novo or inherited rare variants in half (16/32) of ASD families which all probands have normal or VOUS (Variant of Uncertain Clinical Significance) CMA results. In this study, after screening the chromosome structural abnormalities of a proband affected with ASD by CMA, WES was performed on the patient and the parents, and some rare de novo or inherited rare variants have been detected using trios based bioinformatics analysis. To evaluate the variants in ASD, we rank the variants by SFARI Gene score, HPO (Human Phenotype Ontology) and protein function damaging, manual searching PubMed, and we also validated some of candidate variants in other members in this family by Sanger sequencing. We confirmed a de novo homozygous mutation in SPG11 (p.C209F), two inherited compound heterozygote mutations in SCN9A (p.Q10R and p.R1893H) and BEST1 (p.A135V and p.A297V). We also confirmed heterozygous mutations in TSC1 (p.S487C) and SHANK2 (p.Arg569His) inherited from mother.
At present, 1 % of total population are affected with Autism Spectrum Disorders (ASD) and the number is still rising [1]. Multiple lines of evidence suggested that ASD has a strong genetic background. Around 10 % of ASD can be detected with rare Chromosome structural abnormalities and Copy Number Variations (CNVs)[2]. Chromosomal microarray (CMA) has been suggested for first tier clinical diagnostic test for ASD, and ASD with normal CMA results might also undergo single gene disorders test, for example, genes for Fragile X Syndrome and Rett Syndrom etc.[3]. With High-throughput Sequencing (HTS) widely used in ASD genetic studies[4], hundreds of genes have been found to be associated with ASD [5]. Nowadays, some rare penetrant ASD genes related therapeutic methods are being developed and sound promising[5]. Whole Exome Sequencing (WES) has been widely used for diagnosis in rare disorders or some diseases with unknown genetic basis[6]. In our previous work, trios (father, mother and proband) based WGS (Whole Genome Sequencing) were used to reveal clinical relevant de novo or inherited rare variants in half (16/32) of ASD families which all probands have normal or VOUS (Variant of Uncertain Clinical Significance) CMA results. A recently published study also showed that the molecular diagnostic yield of CMA plus WES for diagnosis of ASD is 15.8 % in 95 trios[7].
In this study, we firstly screened the chromosome structural abnormalities in this proband affected with ASD by CMA, but, no clinical relevant copy number gain or loss was found. Afterwards, we performed WES of the proband and the parents. Some candidate rare de novo or inherited rare variants have been detected using trios based bioinformatics analysis, and then we ranked the variants according to SFARI Gene score[8], HPO (Human Phenotype Ontology), protein function damaging, and manual searched PubMed for previous reported studies, and finally we validated the candidate variants in this family by Sanger sequencing. We confirmed a de novo homozygous variants in SPG11(p.C209F), two inherited compound heterozygote mutations in SCN9A (p.Q10R and p.R1893H) and BEST1 (p.A135V and p.A297V), and two rare maternal inherited heterozygous variants in TSC1 (p.S487C) and SHANK2 (p.Arg569His). These variants detected by trio WES might explain phenotype of the proband in this family.
The proband (III-1) is a 2.5-year-old boy with ASD, who was delivered preterm at gestational age of 34 weeks by cesarean section with premature rupture of membranes (PROM) and low birth weight (2.5 kg). The boy might be delivered with early intrauterine infection as the concentration of C-reactive protein (CRP) was 9mg/L and 54 mg/L at day one and three after birth respectively. Note that normal CRP level is < 8 mg/L. CRP level dropped to < 1 mg/L by intravenous injection of antibiotic Azlocillin and Rocephin at day six after birth.
Abnormal communication behavior was observed at 1.5 year old, such as no verbal or body language communication, no eye contact, no response to name calling, and lacking of parental attachment. The restricted and repetitive behaviors can also be observed, such as walking in circles, watching TV ads and no animation, closing the door, and walking on tiptoe. Some other clinical features include normal hearing, broken palmprint in right hand, boundaries of EEG (Electro Encephalo Gram), less sleep and so on.
Informed consent was obtained for all participated family members. This study was performed in accordance with the Declaration of Helsinki protocols and was approved by the Ethics Committee of Shenzhen Children’s Hospital. The proband and their relatives were interviewed to infer a comprehensive clinical history in order to rule out Autism Spectrum Disorder.
Sample collection, Chromosome Microarray (CMA) and Whole Exome Sequencing3-5 ml Peripheral blood was drawn from the proband, parent and other members. Genomic DNA was extracted using Magbind Blood DNA Kit (CW Biotech, Beijing, China) according to manufacturer’s instructions and stored in a -20 ℃freezer . Chromosome Microarray (CMA) was performed by CytoScan® HD array kit (Thermo Fisher, Hudson, New Hampshire, USA) run in GeneChip® System 3000Dx (Thermo Fisher, Hudson, New Hampshire, USA). Whole Exome was captured using SureSelect Human All-Exon V5 (Agilent, Santa Clara, CA, USA) and sequenced by Illumina Hiseq X-ten (Illumina, San Diego, CA, USA) at paired-end 150bp mode. Expected 10 G raw data (or 100 X depth) were generated for each sample.
Rare De novo and inherited callingFirstly, Clean reads were generated by moving remove adapter (GATCGGAAGAGCACACGTCT and AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT) and filtered out read with low quality by Trimmomatic[9], and clean read were aligned to human reference (Hg19) by BWA-MEM[10]. and then variants calling were performed by GATK [11], and finally the variants were annotated by ANNOVAR [12]. For de novo and inherited variants calling from proband-father-mother trios, we used algorithm from our previous study[13] and detailed in the main text.
Rare variants prioritization and phenotype evaluationFirstly, we filtered out the variants with MAF (Minor Allele Frequency) < 0.05 in 1000 genome or our inhouse database; secondly, we filtered candidate autism genes mutation according to autism genes lists downloaded from SFARI gene database[8] on Feb. 28, 2016, we used S-symdromic gene, 1-high confidence gene, 2- srong candidate gene, 3- suggestive evidence gene, and 4-minimal evidence gene as input; thirdly, we used HPO (Human Phenotype Ontology) term (autism or phenotype similar to autism) to predict the gene that might related to autism according to phenotype; finally, we made variants prioritization according to protein conservation and damaging prediction score by SIFT [14] and PolyPhen-2[15], etc.
Sanger sequencing to confirm candidate variants in the familyPCR (Polymerase Chain Reaction) was performed in ABI9700 PCR system (Thermo Fisher, Hudson, New Hampshire, USA) using primer listed in Supplementary Table 1, and the PCR product was sequenced in ABI 3730xl sequencer (Thermo Fisher, Hudson, New Hampshire, USA).
Chromosomal microarray (CMA) can detect Copy Number Variations (CNVs) and has been suggested for first tier clinical diagnostic test for ASD. In the study, no clinical relevant copy number loss or gain was found in the proband.
Whole Exome Sequencing and Variants prioritizationWhole Exome Sequencing was performed on the father-mother-proband trio at averaged 100 fold depth, and the de novo and inherited variants are called from this trio (father-mother-proband) according to our previous study[13]. We used a pipeline to identify the candidate variants (see Fig. 1). Candidate variants identified in the proband are listed in Table 1.
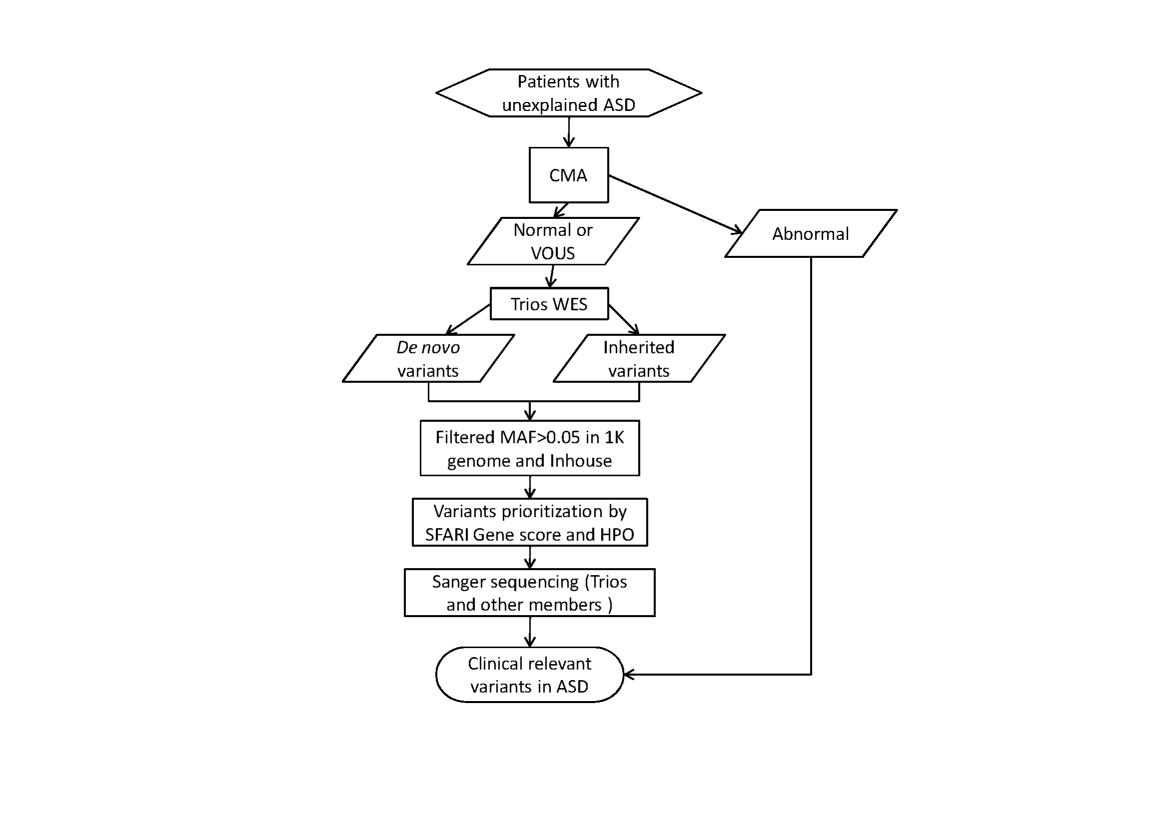 Fig.1 Flowchart to Detect Rare Relevant Variants in Suspected ASD Patient. Abbreviation: WES, Whole Exome Sequencing, VOUS Variant of Uncertain Clinical Significance, HPO, Human Phenotype Ontology; After screening the chromosome structural abnormalities by CMA, WES was performed on the patient and the parents, rare (MAF ≤ 0.05) de novo or inherited were ranked by SFARI Gene score, HPO (Human Phenotype Ontology) and protein function damaging, Sanger sequencing and PubMed were used to identify clinical relevant variants in ASD.
Fig.1 Flowchart to Detect Rare Relevant Variants in Suspected ASD Patient. Abbreviation: WES, Whole Exome Sequencing, VOUS Variant of Uncertain Clinical Significance, HPO, Human Phenotype Ontology; After screening the chromosome structural abnormalities by CMA, WES was performed on the patient and the parents, rare (MAF ≤ 0.05) de novo or inherited were ranked by SFARI Gene score, HPO (Human Phenotype Ontology) and protein function damaging, Sanger sequencing and PubMed were used to identify clinical relevant variants in ASD.
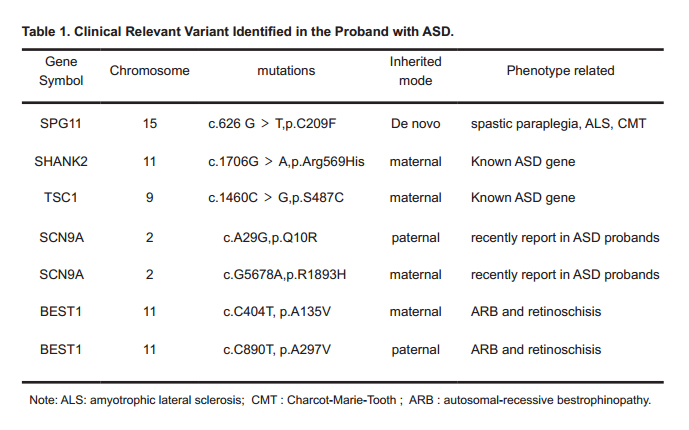 Table 1. Clinical Relevant Variant Identified in the Proband with ASD.
Table 1. Clinical Relevant Variant Identified in the Proband with ASD.
We used Sanger sequencing to confirm the variants in the trio and other members that did not undergone the WES. We confirmed a de novo homozygous variants in SPG11 (p.C209F) from the proband (III-1). We also confirmed compound heterozygous mutations in SCN9A (p.Q10R and p.R1893H) in the proband, and proband’s mother (II-1) and father (II-2) were carriers for heterozygous mutations in SCN9A (p.Q10R) and SCN9A (p.R1893H), respectively. Further validation confirmed that maternal grandfather (I-1) and grandfather (I-2) were carriers for heterozygous mutations in SCN9A (p.R1893H) and SCN9A (p.Q10R), respectively. Another compound heterozygous mutations in BEST1 (p.A135V and p.A297V) have also been confirmed, heterozygous mutations in BEST1 (p.A135V) and BEST1 (p.A297V) are inherited from proband’s mother (II-1) and father (II-2), respectively. Interesting, we identified two heterozygous mutations in SHANK2 (p.Arg569His), TSC1 (p.S487C), which are inherited from Proband’s mother (II-1). All results are shown in Fig.2.
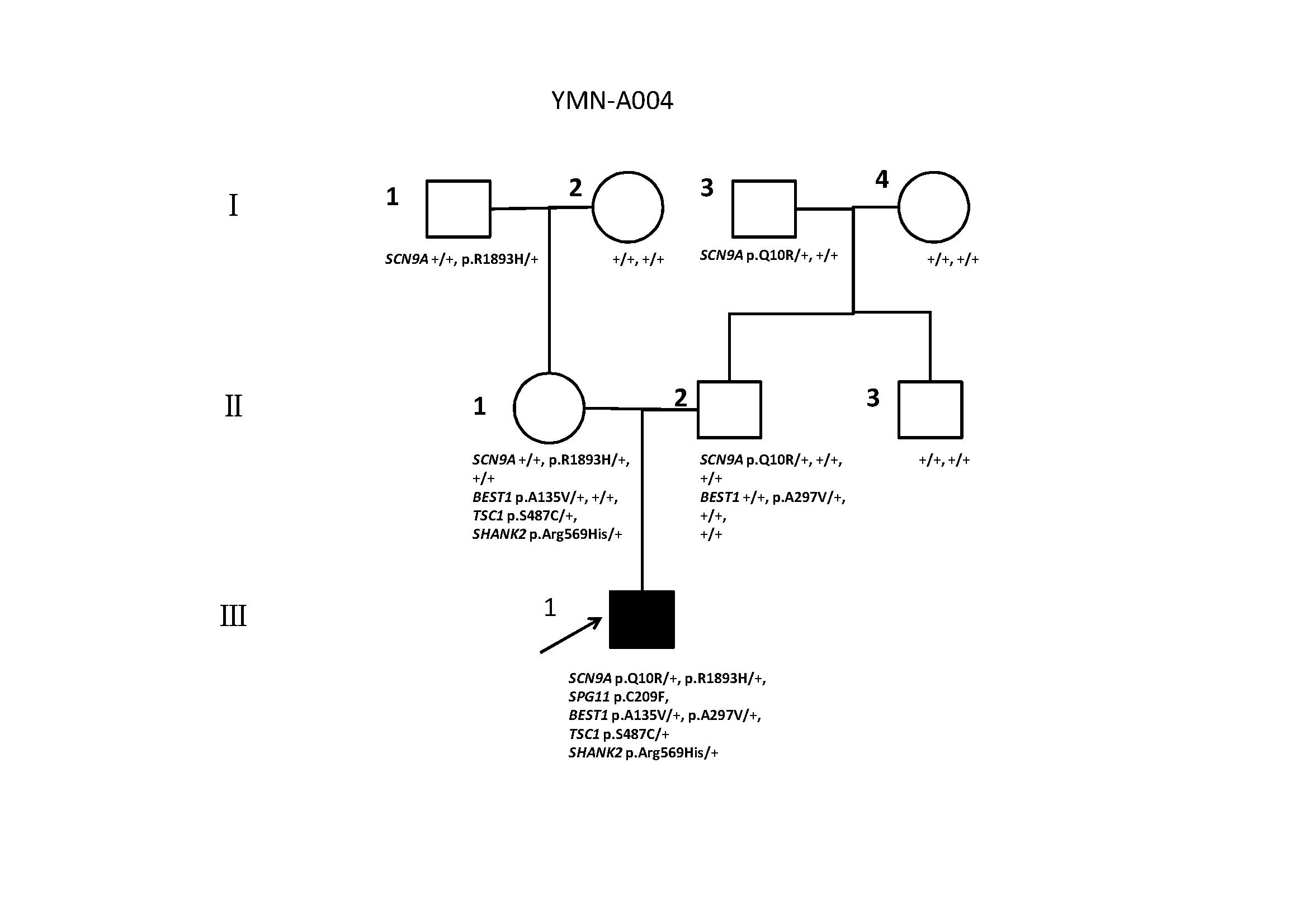 Fig. 2. Clinical Relevant Variants Detected in this Family.
Fig. 2. Clinical Relevant Variants Detected in this Family.
Males are denoted by squares and females by circles. Black symbols indicate individuals diagnosed with ASD, open symbols denote unaffected individuals, and arrows indicate ASD proband in this family; II-1, II-2, and III-1 are trios for WES (whole exome sequencing), and “+” indicates the allele containing the reference; “+/+” for genes on the autosomal chromosome or the X chromosome in female; SCN9A p.Q10R/+, SCN9A p.R1893H/+, SPG11 p.C209F, BEST1 p.A135V/+, BEST1 p.A297V/+, TSC1 p.S487C/+, SHANK2 p.Arg569His/+ and confirmed by Sanger sequencing in II-1, II-2, and III-1, and only SCN9A p.Q10R/+ and SCN9A p.R1893H/+ are confirmed in other family members.
With CMA recommended to be used for first tier genetic diagnosis for ASD[3], lots of clinical relevance chromosomal abnormalities have been identified, but the diagnosis yield (around 10 %) is still low, and lots of chromosomal abnormalities have been annotated with VOUS (Variant of Uncertain Clinical Significance)[2]. With High Throughput Sequencing (HTS) widely used in ASD genetics studied, lots of ASD relevant genes have been identified[4, 5]. We have identified clinical relevant variants in 50 % of Canadian ASD families with negative CMA results by Whole Genome Sequencing (WGS)[13]. This might suggest that HTS can be used as further screen of clinical relevant variants in ASD patient with normal or VOUS results. In the current study, copy number loss has been identified in 15q (q11.1-q11.2), totaling 2.209 Mb, and annotated with VOUS (CMA providers). So we ordered WES for the proband and his parents. After a systematically identification of de novo and rare inherited variants in the family, we used the updated SFARI Gene and HPO database, damaging prediction software, MAF, to rank the variants. After manually searching the PubMed, we confirmed a de novo homozygous mutation in SPG11 (p.C209F), two inherited compound heterozygote mutations in SCN9A (p.Q10R and p.R1893H) and BEST1 (p.A135V and p.A297V), and two rare maternal inherited heterozygous mutations in TSC1 (p.S487C) and SHANK2 (p.Arg569His) in this proband.
TSC1 and SHANK2 are frequently reported genes related to Autism, or known autism genes. TSC1 (p.S487C) leads to the substitution of Ser to Cys at amino acid 487 in exon 15. TSC1 has been related to syndromic autism, where a small subpopulation of individuals will develop autism. The SFARI gene score of TSC1 is “S”, which means “syndromic”. Moreover, a loss of function mutation in the TSC1 gene results in Tuberous sclerosis[16]. And about 25 % to 60 % of the Tuberous sclerosis patients have autism[17]. SHANK2 (p.Arg569His) leads to the substitution of Arg to His at amino acid 569. The SFARI gene score of SHANK2 is “2”, which means “strong candidate”. SHANK2 is a multi-domain scaffolding protein that all localize to postsynaptic sites of excitatory synapses in the brain[18]. Several studies have identified rare mutations in the SHANK2 gene in autistic and intellectually disabled individuals[19]. SHANK2-/- mice showed that SHANK2 seemed to serve different interrelated functions in glutamate receptor targeting/assembly which can lead to the core symptoms of ASD[20]. The Mutation Taster predicted that the two mutations have the disease causing mutations whereas Polyphen-2 and SIFT suggested that the substitutions are probably damaging or damaging which means that the proteins have a high probability of being affected by the mutations. In addition, Sanger Sequencing analyses of the parent revealed that only his unaffected mother has the two heterozygous mutations (Fig. 2). Though low penetrance does exist in autism related genes[21], our results may not support that TSC1 (p.S487C) and (p.Arg569His) explain the ASD phenotype in this proband.
SCN9A might be a novel Autism related gene. SCN9A, encoding sodium voltage-gated channel alpha subunit 9 or Na(v)1.7, is firstly reported as a gene related to pain disorders[22]. SCN9A mutation can cause insensitive to pain and inherited erythromelalgia[23], and then SCN9A mutations is also reported as genetics causes for febrile seizures and Dravet syndrome[24, 25]. Since epilepsy and ASD can co-occur frequently, some epilepsy gene are strong candidate for ASD[26]. This may suggest epilepsy and ASD may share common biology. Yuen et al. identified the same mutation in SCN9A (p.V205fs) in two sibling affected with ASD[27]. We confirmed a compound heterozygote mutations in SCN9A (p.Q10R and p.R1893H) in the proband (III-1). Proband’s mother (II-1) and maternal grandfather (I-1) are carriers for SCN9A (p.R1893H). Proband’s father (II-2) and grandfather (I-2) are carriers for SCN9A (p.Q10R). Our results might give a clue that SCN9A might play a role in ASD, but further studies may be necessary to confirm SCN9A’s role in ASD.
We also confirmed rare disease genes mutated in the proband, such as BEST1 and SPG11. BEST1, encoding bestrophin 1, is a gene whose mutation causes autosomal-recessive bestrophinopathy (ARB)[28] and retinoschisis[29]. We confirmed a compound heterozygous mutation in BEST1 (p.A135V and p.A297V) in the proband, and BEST1 (p.A135V) and BEST1 (p.A297V) are inherited from proband mother (II-1) and father (II-2), respectively. SPG11, encoding spastic paraplegia 11, is a gene whose mutations can cause spastic paraplegia[30], juvenile amyotrophic lateral sclerosis (ALS)[31] and Charcot-Marie-Tooth disease[32]. We confirmed a de novo mutation in SPG11 (p.C209F) from the proband (III-1). Further clinical evaluation associated with this two genes related disorder would be necessary to confirm these disorders.
We performed WES to the proband-father-mother trio, bioinformatics analysis and Sanger sequencing confirmed two heterozygous mutations in two known ASD genes, SHANK2 (p.Arg569His) and TSC1 (p.S487C) inherited from proband’s mother (II-1). We also confirm a compound heterozygote mutation in SCN9A (p.Q10R and p.R1893H) in the proband (III-1), and a gene recently reported in ASD patients. We also rare disease related mutation, heterozygous mutations BEST1 (p.A135V and p.A297V) and de novo mutation in SPG11 (p.C209F). Although single proband and lack of functional study, our results might provide a clue for others to identify ASD related mutations.
We are sincerely grateful to the family members for their participation and cooperation in the study. The authors would like to thank the groups that generated Whole Exome Sequencing data from Joint lab between Children’s Hospital of Fudan University and Wuxi NextCODE Co., Ltd. This project was support by Shenzhen Science Technology Research & Development Fund (Program No. :JCYJ20130401114111453, JCYJ201404111 50159429, JCYJ20140416141331514 and JCYJ20140415151845360) from Shenzhen Science Technology and Innovation Commission, Longgang District Science Fundamental Research fund (Program No.: 201406063001021) from Longgang District Science Technology and Innovation Commission of Shenzhen, and National High Technology Research and Development Programs (“863 Program”) (No. 2015AA020405) and by Shenzhen Imuno Biotech Co., Ltd (Program No.: 2015A004).
J. Z., R.X., M.B. and S. Z. planned and coordinated the project, M.W, J. L. and R.X. wrote the manuscript; X. L. prepared samples and extracted DNA; Y.X. performed CMA, F.H. and Y.W. conducted WES and bioinformatics analysis. These authors contribute equally to this manuscript.
Mingbang Wang and Yan Wang received honoraria and research support from Shenzhen Imuno Biotech Co., Ltd. But this does not alter the authors' adherence to Journal policies on sharing data and materials. There is no others competing interests to declare.
Supplementary Fig. 1: CMA results for the proband.
Supplementary Table 1: Primers for validation candidate variants.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
Zhou J, Yang F, Li X, Zhou S, He F, Liang J, Wang Y, Wang M, Xu R. Whole Exome Sequencing Revealed Clinical Relevant Variants in A Family Affected with Autism Spectrum Disorder. J Psychiatry Brain Sci. 2016;1(4):3; https://doi.org/10.20900/jpbs.20160017

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




