Location:Home >> Detail
Med One. 2016; 1: e160022. https://doi.org/10.20900/mo.20160022

1 Department of Biomedical Engineering, Tianjin University, Tianjin 300072, P.R. China
2 Department of Radiology and Imaging Sciences, NIH, Bethesda 20852, USA
3 Department of Genomics Research, R&D Solutions, Elsevier Inc., Rockville, MD 20852, USA
4 Unit on Statistical Genomics, NIMH/NIH, Bethesda 20852, USA
*Corresponding Author: Dr. Hongbao Cao, Department of Genomics Research, R&D Solutions, Elsevier Inc., Rockville, MD 20852, USA. Tel: 240-461-9642.
Background: Previous studies have shown that Helicobacter pylori infection (HPI) is related to a reduced risk of esophageal adenocarcinoma (EAC) by unknown biological mechanisms. It is hypothesized that EAC and HPI have strong genetic associations.
Methods: An integrated analysis, using large-scale ResNet relation data and gene expression data for HPI and EAC, to identify potential EAC risk genes from a HPI-gene group was conducted. Disease-gene relation data were acquired from the Pathway Studio ResNet Mammalian database. Gene expression data were acquired from samples of 92 subjects including 64 EAC cases and 28 normal controls.
Results: Genes linked to HPI and EAC present significant overlap (79 genes, p-value = 2.5E-75) and play roles within multiple common genetic pathways (enrichment p-value ≤ 5.05E-17 for the top 10 pathways) that are implicated with both diseases. A genetic network of 32 genes was identified through which HPI may exert influence on EAC. There were 6 HPI genes that presented significant differences (p-value < 1e-10) between EAC cases and controls, including: MUC13, AQP3, TFF3, SFTPD, NOD2, and PIGR. Network analysis showed that these genes demonstrated strong functional associations with EAC and may be potential EAC risk genes.
Conclusion: Results from this study support the hypothesis that complex genetic associations exist between HPI and EAC, and that HPI-related genes may also play roles in EAC pathogenic development. This provides new insights into EAC candidate gene identification.
Esophageal adenocarcinoma (EAC) is a rapidly increasing incidence, high-mortality cancer in developed countries [1]. Studies suggest that at least 95 % of EAC cases arise from a metaplastic condition known as Barrett's esophagus [2]. Genetic studies using genome-wide association study (GWAS) and GED have been conducted to explore the genetic risks associated with EAC [3, 4]. Hundreds of EAS-linked genes have been reported. The basic carcinogenesis mechanisms underlying EAC clinical outcomes remain unclear. Genetic associations between Helicobacter pylori infection (HPI) and EAC were studied here in order to better understand the genetic bases of EAC, and identify novel potential genes for it.
Helicobacter pylorus is a gram-negative bacillus usually found in human gastric mucosal epithelium. Affecting over half of the world's population, HPI is a cause of gastroesophageal reflux disease (GERD) and a risk factor for GC [5]. HPI seems to associate with a reduced EAC risk. People with HPI have a greater than 40 % lower incidence of EAC than those without [6, 7]. Biological explanations for this HPI protective effect in the case of EAC remains unclear. It is believed that the reduced risk may be linked to lower gastric acid levels in HPI patients [7, 8].
In recent years, the Pathway Studio ResNet database has been widely used to study modeled-relationships between proteins, genes, complexes, cells, tissues, and disease [9]. This study integrated large-scale ResNet relation data and gene expression data to test the hypothesis that HPI and EAC share a genetic base, and that HPI-related genes may also associate with EAC. The results support the HPI-EAC correlation hypothesis and may identify potential novel risk genes for EAC.
Large scale HPI-gene and EAC-gene ResNet relation data were studied to identify shared genes and genetic pathways. Integrated EAC expression data was examined to identify novel genes from the HPI-gene group. Lastly, a functional network analysis was performed to study any potential pathogenic significance of these EAC-candidate genes.
2.1 HPI-Gene and EAC-Gene data acquisitionDisease-gene relation data for HPI and EAC were acquired from the Pathway Studio ResNet relation database. It has been widely used to study modeled relationships between proteins, genes, complexes, cells, tissues, and diseases (http://pathwaystudio.gousinfo.com/Mendeley.html). It is updated weekly and is the field’s largest database [10]. In addition to the complete gene lists of genes, supporting references for each disease-gene relation appear in Supplementary Tables S1 and S2, and include reference titles and the related sentences where these relations were identified. This information could be used to located detailed descriptions of how a candidate genes relate to HPI and/or EAC.
2.2 Identification of risk genesA gene expression data set (GSE13898) of 92 subjects was used to test genes related to HPI which have not been reported to associate with EAC. This was to identify potential EAC risk genes.
The gene expression profiles acquired from 64 primary esophageal adenocarcinoma, 15 Barrett's esophagus, and 28 surrounding normal fresh frozen tissues were used for the microarray. All tissues were obtained after curative resection following pathologic confirmation at the University of Texas MD Anderson Cancer Center (MDACC). Microarray experiment and data analysis were done in the Department of Systems Biology at MDACC. Raw and processed data were deposited in NCBI GEO Datasets, which are available online at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE13898.
2.3 Network analysis of EAC risk genesA network analysis between 6 target genes and EAC was performed to identify any entities that could act as a bridge connecting the gene and EAC. This was done to validate potential candidate EAC risk genes. Target entity analysis included proteins/genes, small molecular/drugs, and functional classes. The relation data between these target entities and the 6 target genes and EAC were acquired from Pathway Studio ResNet database for analysis.
A systematic analysis of the HPI-Gene and the EAC-Gene ResNet relation data to identify genes associated with HPI and EAC was conducted. Results showed that 276 genes associated with HPI. This is supported by 720 scientific references between 1992 and June 2016 (Supplementary Tables S1a and S1b). For EAC, 293 genes, supported by 700 references between 1993 and June 2016 (Supplementary Tables S2a and S2b) were identified. A significant overlap of 79 genes between HPI-genes and the EAC-genes (Right tail Fisher’s Exact test, p-value = 2.5E-75), as shown in Fig. 1 (see Supplementary Tables S3a and S3b for the gene list and references) exists.
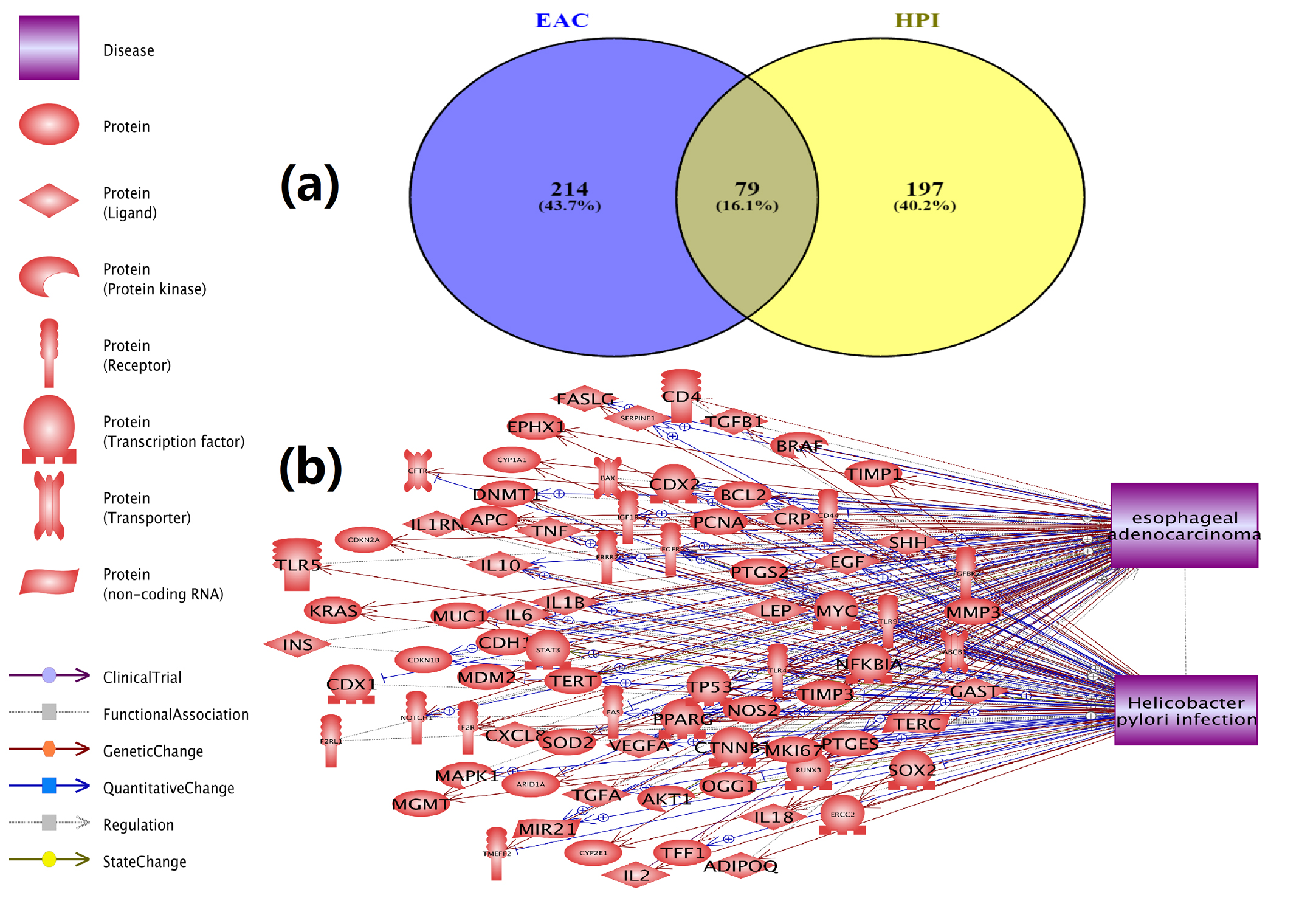 Fig. 1 Genetic association between HPI and EAC.
Fig. 1 Genetic association between HPI and EAC.
(a) Venn diagram for HPI-genes and EAC-genes; (b) The 79 genes linked with both HPI and EAC.
A Pathway Enrichment Analysis (PEA) using Pathway Studio was conducted to test the functional profile of the 79 genes associated with both HPI and EAC.
The 10 most significantly-enriched pathways (p-value ≤ 5.05E-17) appear in Table 1. A total of 637 pathways/gene sets were enriched with p-value < 1e-3 including 77 of the 79 genes (Supplementary Table S4).
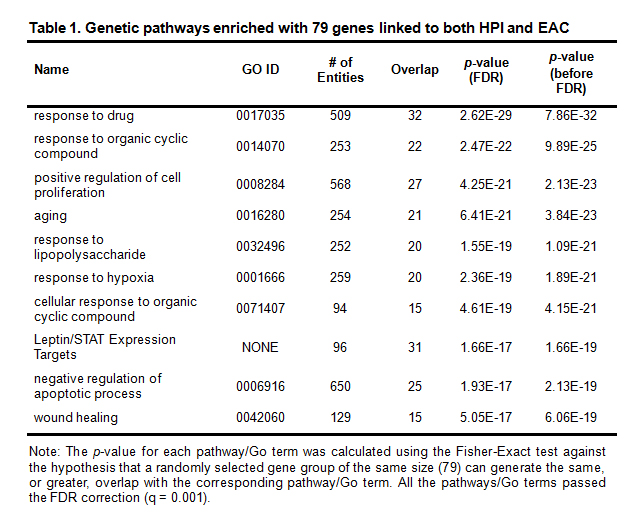 Table 1. Genetic pathways enriched with 79 genes linked to both HPI and EAC
Table 1. Genetic pathways enriched with 79 genes linked to both HPI and EAC
Note: The p-value for each pathway/Go term was calculated using the Fisher-Exact test against the hypothesis that a randomly selected gene group of the same size (79) can generate the same, or greater, overlap with the corresponding pathway/Go term. All the pathways/Go terms passed the FDR correction (q = 0.001).
PEA results showed: 37 pathways/gene sets (57 unique genes) related to cell growth and proliferation; 34 (49 unique genes) to cell apoptosis; 10 (29 unique genes) to protein kinase; 9 (26 unique genes) to protein phosphorylation; 9 (38 unique genes) to transcription factors; 6 (30 unique genes) to immune system; and, 2 (21 unique genes) to single-organism developmental process.
Many of these pathways have been implicated in both HPI and EAC. These include: the response to lipopolysaccharide (GO ID: 0032496) [11, 12]; ageing (GO ID: 0016280) [13, 14]; response to hypoxia (GO ID: 0001666) [15, 16]; and, positive regulation of cell proliferation (GO ID: 0008284) [17, 18]. The data for these significantly enriched pathways appears in Supplementary Table S4.
The results suggest that HPI and EAC share multiple genetic pathways. It is through these shared pathways that a large number of genes play roles affecting the pathogenic development of both diseases.
3.2 Possible co-regulations between HPI and EACFurther functional network analysis, using PS, showed that, 32 of 79 genes are downstream targets of HPI (influenced by HPI), while also being an EAC upstream regulator (Fig. 2). HPI may influence EAC pathogenic development through the regulation of these 32 genes. For each relation (shown by an arrow) in Fig. 2, there is support from one, or more, references (Supplementary Table S3b), which could be used for a detailed description of each relation.
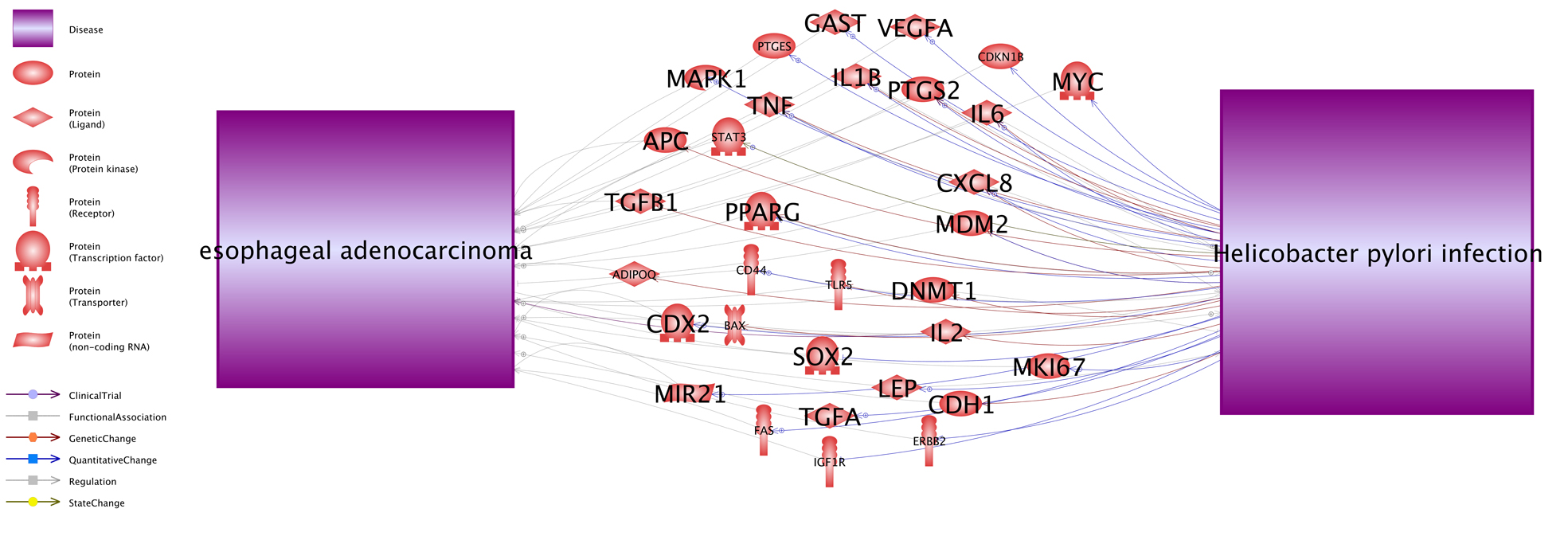 Fig. 2 A HPI→Gene→EAC pathway contain 32 genes.
Fig. 2 A HPI→Gene→EAC pathway contain 32 genes.
Networks were generated using the ‘network building’ module of Pathway Studio. The definition of the entity types and relation types in the figure can be found at http://pathwaystudio.gousinfo.com/ResNetDatabase.html
The results suggest that any gene linked to HPI may be worthy of study for its potential relation to EAC. These genes affect the HPI pathogenic development, which in turn may influence the disease status of EAC.
3.3 Expression analysis HPI-genesThe ResNet relation data analysis showed that more HPI genes were not linked to EAC than these were (197 vs. 79; see Fig. 1). A gene expression analysis was conducted to study expression differences between EAC cases and controls for these 197 genes in order to identify those linked to HPI which were also potential EAC risk genes. Fig. 3 provides the ‘–log10’ transferred p-values (q = 0.001 for FDR) of each gene.
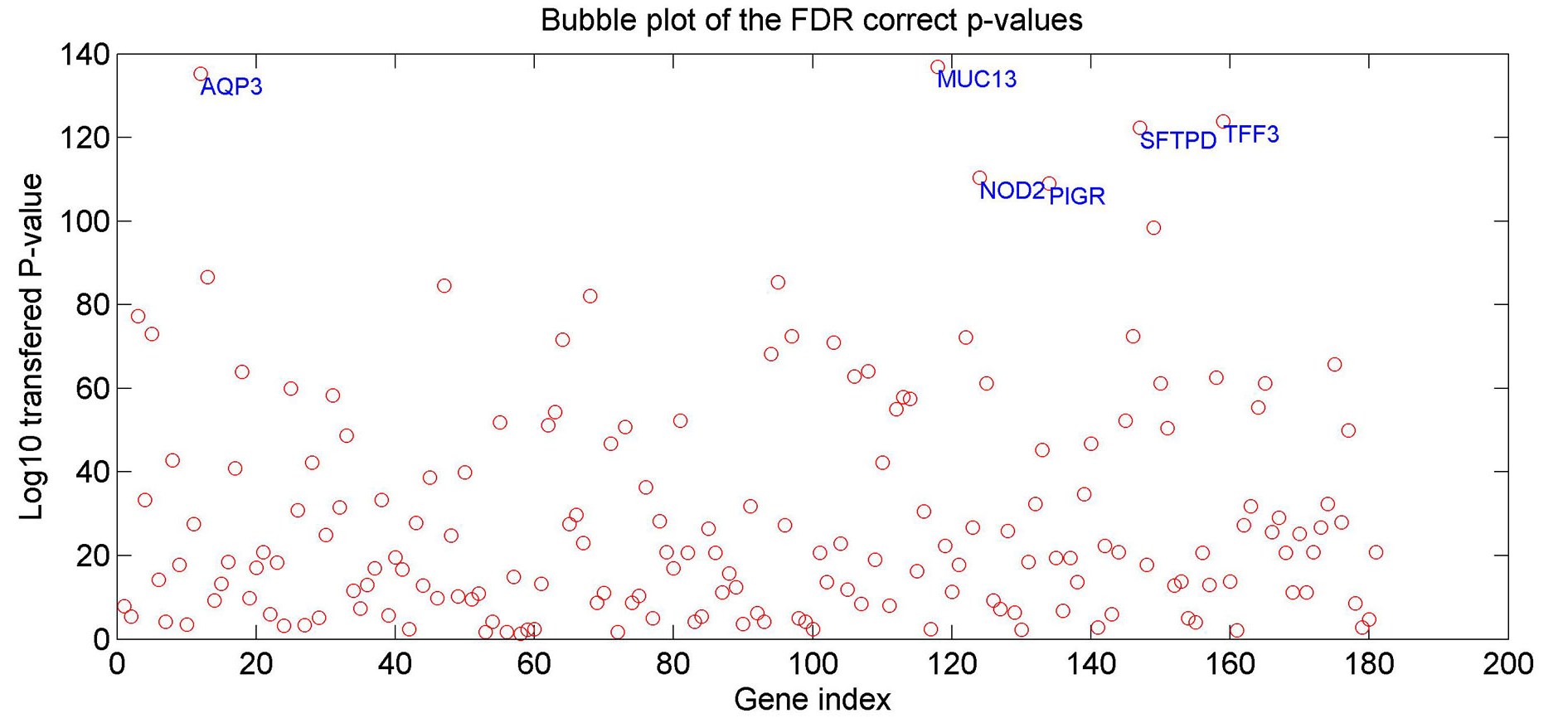 Fig. 3 The p-values for the 197 HPI genes for EAC case/control expression comparison.
Fig. 3 The p-values for the 197 HPI genes for EAC case/control expression comparison.
The p-values have been through FDR correction with q=0.001 and logic transformation using ‘–log10’. The six genes demonstrating significant differences (p-value < 1e-10) appear at their corresponding positions.
In the gene expression analysis, 62 of 197 HPI genes passed the FDR correction (q = 0.001. See Supplementary Table 5). Six genes presented a significant difference (p-value < 1e-10) between EAC cases and controls. These were: MUC13, AQP3, TFF3, SFTPD, NOD2, and PIGR. According to the PS ResNet database, these 6 genes presented no direct relation with EAC in that there was no reference reporting an association between these genes and EAC. However, they demonstrate strong indirect linkage to EAC, bridged by 29 genes/proteins, 10 small molecular, and 7 functional classes (see Fig. 4). The 46 entities and the 141 relations with 1,385 supporting references in Fig. 4 appear in Supplementary Table S5a and S5b, respectively.
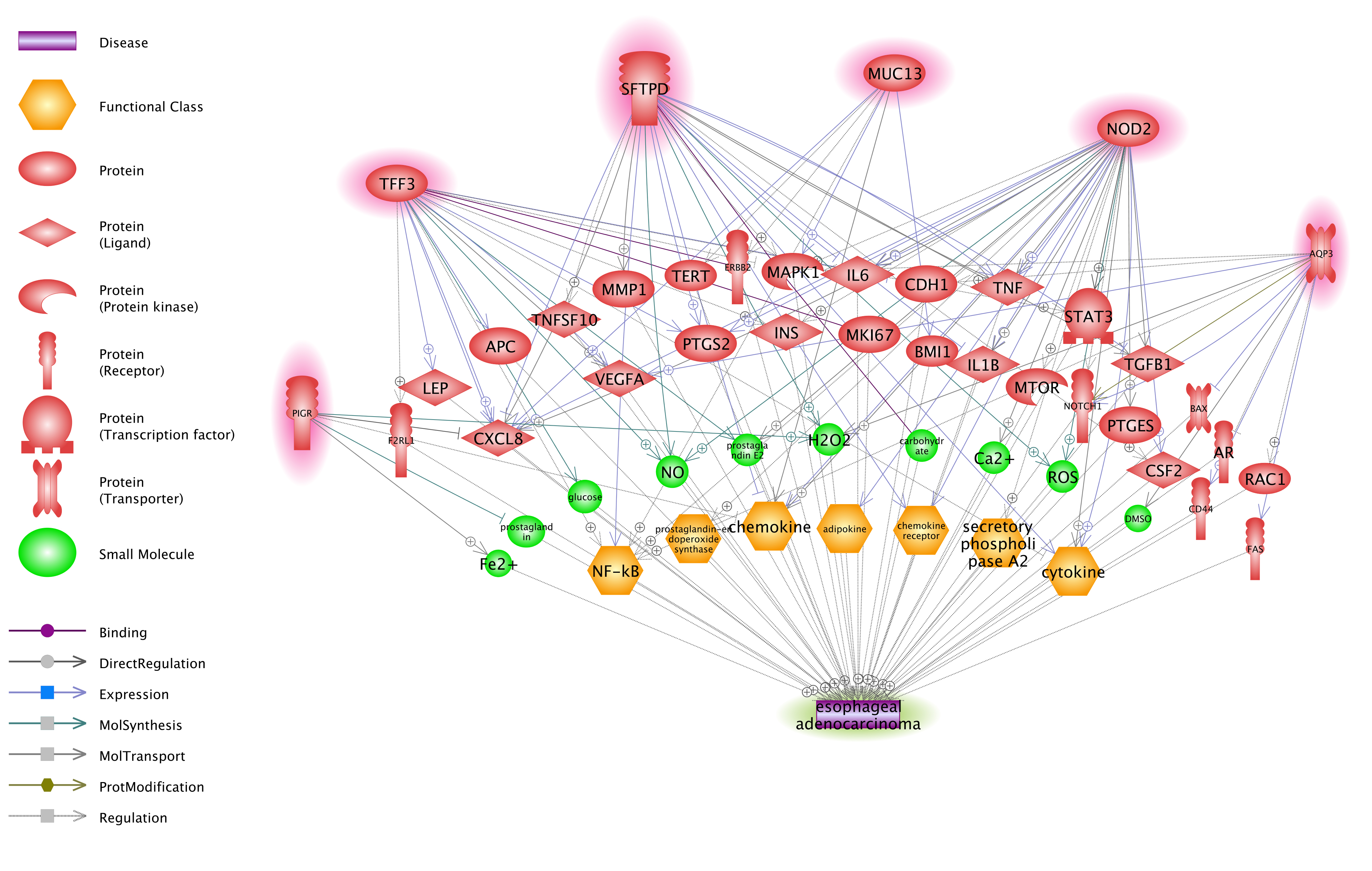 Fig. 4 Functional network between 6 HPI genes and EAC.
Fig. 4 Functional network between 6 HPI genes and EAC.
The network was constructed with ‘network building’ module of Pathway Studio.
Previous studies showed that HPI is strongly linked to reduced EAC incidence via an unclear mechanism [6, 7, 19]. This study used large-scale ResNet relation data and gene expression data to study shared genes and genetic pathways between HPI and EAC. The approach identified potential novel EAC risk genes.
The results showed that genes linked to HPI and EAC present significant overlap (79 genes, p-value = 2.5E-75). All but 2 (77 of 79) genes were significantly enriched within 637 pathways (p-value < 1e-3, FDR corrected: q = 0.005). Many of these pathways have been linked to both HPI and EAC. They include: response to lipopolysaccharide (GO ID: 0032496); ageing (GO ID: 0016280); response to hypoxia (GO ID: 0001666); and, positive regulation of cell proliferation (GO ID: 0008284) [11-18]. These results suggest that HPI and EAC share multiple genetic pathways. A large number of genes regulate both diseases pathogenic development through these pathways.
A 32-gene network was discovered through which HPI may affect the disease status of EAC (Fig. 2). These findings provide further support for the hypothesis that HPI genes may regulate EAC pathogenic development.
A closer study of the 197 HPI only genes (Fig. 1 (a)) using EAC gene expression data showed that a large portion (62/197 = 31.47 %, q = 0.001 for FDR) of these HPI genes also demonstrated differences between EAC cases and controls (FDR corrected p-value < 0.001) (Fig. 3). Six genes were identified as potential EAC markers (FDR corrected p-value < 1e-10), including: MUC13; AQP3; TFF3; SFTPD; NOD2; and, PIGR. Further validation using a ResNet network analysis showed that these six genes presented strong indirect correlation with EAC forming a functional genetic network supported by 1,385 supporting references (Fig. 4). Through this network, multiple pathways could be identified through which a gene may affect EAC disease status. One example, NOD2, has been reported to be involved in the production of microbicidal reactive oxygen species (ROS) [20], which play an important role in EAC development [21]. This finding supports a NOD2 → ROS → EAC pathway. Another possible MUC13 → EAC pathway was identified. MUC13 has been shown to regulate chemokine secretion [22]. Chemokine receptors are Class A GPCRs coupled with Gαi heterotrimeric G proteins and play a pivotal role in EAC tumorigenesis and metastasis [23]. By regulating chemokine secretion, MUC13 may regulate EAC pathogenesis through a chemokine pathway which would build a MUC13 → chemokine pathway → EAC regulation mechanism.
In conclusion, the results from this study support the hypothesis that HPI and EAC present significant genetic level associations, which may explain their clinical correlations. Moreover, novel potential EAC genes can be identified by integrating ResNet relation data and gene expression data. This is the first study that we know of that integrates large-scale ResNet relation data and gene expression data to study molecular associations between HPI and EAC. The findings of this study may provide new insights into the current field of HPI-EAC correlation study and warrants further study using more data sets to identify novel potential EAC risk genes.
We would like to thank Dr. Sana Khan for her suggestions and writing help in the development of this manuscript. Dr. Khan is with Department of Genomics Research, R&D Solutions, Elsevier Inc.
The author HC is with Elsevier Inc.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
Supplementary File 1: Excel File
Zhou P, Foster P, Cao H. Cross-Disease Analysis Reveals Novel Risk Genes for Esophageal Adenocarcinoma. Med One. 2016 Oct 25; 1: e160022. https://doi.org/10.20900/mo.20160022

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions



