Location: Home >> Detail
Quantum Mater Res. 2020;1:e200003. https://doi.org/10.20900/qmr20200003

School of Chemistry, University of Melbourne, Melbourne, Victoria 3010, Australia
* Correspondence: Alessandro Soncini.
This article belongs to the Virtual Special Issue "Single Molecule Magnets - In Memory of Professor Alessandro Bencini"
We present a computational investigation of the intramolecular exchange coupling in [LnPc2]0 (Ln = Tb, Dy, Ho, and Er) between the Ln3+ 4f electrons and the spin-1/2 radical on the phthalocyanine ligands. A series of ab initio multi-configurational/multi-reference Complete/Restricted Active Space Self-Consistent-Field calculations (CASSCF/RASSCF), including non-perturbative spin–orbit coupling, were performed on [LnPc2]0 and on the smaller model compound [LnPz2]0. We find that the exchange coupling mechanisms are restricted by symmetry, but also dependent on the spin polarization effect triggered by the Pc2 ligands π–π∗ excitations. The calculated exchange splittings are small, amounting to at most a few cm–1, in disagreement with previous literature reports of strong antiferromagnetic coupling, but in good agreement with recent EPR experiments on [TbPc2]0. Furthermore, the coupling strength is found to decrease from [TbPc2]0 to [ErPc2]0, with decreasing number of unpaired electron spins in the lanthanide ground (Hund’s rule) Russell–Saunders term.
SMMs, Single-Molecule Magnets; CASSCF, Complete Active Space Self-Consistent Field; RASSCF, Restricted Active Space Self-Consistent Field; Ln, Lanthanide; MOs, Molecular Orbitals; SOMO, Singly Occupied Molecular Orbital; AFM, Antiferromagnetic; FM, Ferromagnetic; NMR, Nuclear Magnetic Resonance; DFT, Density Functional Theory
Single molecule magnets based on the lanthanide phthalocyanine double-decker ([LnPc2]±1/0, Ln = lanthanide, Pc = phthalocyanine) are of particular interest due to their large barrier to magnetic relaxation and high blocking temperatures, especially compared to traditional single molecule magnets based on transition metals [1–7].
LnPc2 comes in a number of oxidation states, one of the most interesting forms being the neutral [LnPc2]0, partly because it has a larger barrier for magnetic relaxation, and also because it can be easily sublimated in ultra-high vacuum (UHV) deposition to fabricate molecular spintronic devices [8–18]. Its interest also stems from the fact that [LnPc2]0 has an unpaired electron in the Pc2 ligand moieties, which has been argued to mediate the exchange coupling between the localized Ln magnetic moment and underlying substrates, such as magnetic thin fimls or carbon nanostructures [13,15,17,18].
Understanding the strength and nature of the exchange coupling between the Ln3+ 4f electrons and the organic radical delocalized over the Pc2 rings thus represents an important task. The first contribution in that direction was made in a study of the temperature-dependent magnetic susceptibility of powder samples of [LnPc2]0 [19,20]. In that work, the authors reported saturated values of χT which were systematically smaller than what is theoretically expected for an independent Ln(III)–radical pair. For example, they recorded the following values of χT (cm3 K mol–1) at 300 K (theoretically expected in parentheses): Tb 9.2 (12.2), Dy 13.0 (14.5), Ho 11.3 (14.4), Er 8.4 (11.9). To explain these results the authors proposed that a strong exchange interaction must exist between the lanthanide and the Pc2 radical. This interaction should be antiferromagnetic and at least as large as room temperature (≈200 cm–1) in order to explain the observed values, which at 300 K are appreciably lower than expected for the uncoupled systems [19,20]. It should be noted that a coupling of that magnitude is unusual for exchange involving 4f electrons. In view of the small overlap between the highly localized 4f orbitals and the magnetic orbital(s) of the exchange partner, a much weaker interaction is expected. This issue was not mentioned by the authors [19,20], and their conclusion that the lanthanide is strongly and antiferromagnetically coupled to the Pc2 radical has been repeated unchallenged in review articles [7,21,22].
Recently, evidence to the contrary was derived from a single-crystal EPR experiment on [TbPc2]0 [23]. The field and angle dependent resonance frequencies were found to be consistent with a small ferromagnetic interaction described by the Ising Hamiltonian
–2Jeff SLnz SPc2z (1)
Here, S Ln denotes an effective spin of 1/2 representing the ground state doublet on Tb, and SPc2 denotes the real spin of the Pc2 radical. The exchange splitting derived from the EPR measurement is Jeff = 0.88 cm–1. Note that the choice of writing the exchange coupling Hamiltonian between two pseudo-spin 1/2 as in Equation 1 implies that 0.88 cm–1 corresponds to the energy gap between the ground ferromagnetic exchange Kramers doublet, and the first excited antiferromagnetic exchange Kramers doublet. It is clear that this small interaction is incompatible with the susceptibility data of Trojan et al. [19,20].
We could find only one other published susceptibility measurement on these systems, namely for [DyPc2]0 [24]. The χT data reported by Branzoli et al. [24] disagree with those of Trojan et al., most significantly in the high temperature region, where χT is substantially higher, reaching a value of 14.6 cm3 K mol–1 at 300 K (albeit not fully saturated), compared to 13.0 in Trojan et al. and 14.54 the expected value for the uncoupled system.
To date, there have been only a few computational studies of the exchange coupling in the [LnPc2]0 series. Damjanović et al., based on a combination of NMR measurements and DFT calculations, suggested a ferromagnetic interaction between Pc2 radical and Ln(III) ion [25]. However they did not report on the magnitude of the interaction. DFT calculations in Ref. [17] revealed a correlation between observed magnetic coupling of [LnPc2]0 to a Ni surface with computed spin polarization in the Ln 5d orbitals. Ref. [18] and the recent work of Pederson et al. [26] report ab initio calculations on [TbPc2]0, similar to those of the present work, but did not take into account the effect of spin polarization in the ligand π system, which we show in the present work to be important.
This paper presents a theoretical and computational investigation of the intramolecular exchange coupling mechanisms within [LnPc2]0 (Ln = Tb, Dy, Ho, and Er) molecules. We find that in the simpler CASSCF calculations where the active space consists solely of seven Ln3+ 4f orbitals and the singly occupied molecular orbital (SOMO) of the molecular ligands, the coupling between lanthanides and the radical is constrained to be ferromagnetic by symmetry, and the exchange strength decreases with increasing atomic number, i.e., Tb > Dy > Ho > Er. On extension of the active space to RASSCF calculations, with the previously explored CASSCF active space determining the RAS2 space, π orbitals in RAS1 and π∗ orbitals in RAS3 space, allowing for at most double excitations (two holes in the RAS1 space and two particles in the RAS3 space), a new antiferromagnetic mechanism based on spin polarization is activated, which reduces the overall exchange coupling constant, which remains however ferromagnetic. The computed exchange splittings are found to be small, of the order of 1–10 cm–1 for all four ions. Our best value for [TbPc2]0 is Jeff = 1.92 cm–1, and is consistent in sign and magnitude with the value of 0.88 cm–1 from EPR experiment [23], thus supporting the interpretation of weak ferromagnetic coupling in [TbPc2]0. These results are at variance with the strong antiferromagnetic coupling suggested in Refs. [19,20].
A series of single point state-averaged CASSCF/RASSCF calculations followed by state-interaction via spin–orbit coupling (RASSI–SO) was carried out using the MOLCAS 8.0 code [27]. ANO-RCC-VDZP and ANO-RCC-VDZ basis sets were used on the lanthanide and the ligand atoms, respectively. Crystallographic structures of [TbPc2]0, [DyPc2]0 and [ErPc2]0 were obtained from the literature [9,28]. The structure of [HoPc2]0, for which no crystallographic data could be found, was formed from [TbPc2]0 by replacing Tb with Ho.
CASSCF calculations were performed on [LnPc2]0, employing Cholesky decomposition of the two-electron integrals (with a threshold of 10–6 Eh). The CASSCF active space contains the seven 4f orbitals of the central lanthanide, which transform as b2 + e1 + e2 + e3 in the approximate D4d point group, and the π-SOMO (having a2 symmetry) [29] of the Pc2 rings (see Figure 1, top). RASSCF calculations were performed on a simplified model structure in order to reduce computational cost. The eight outer benzene rings of Pc2 were removed and the remaining structure was adapted to perfect D4d symmetry. The resulting structure, referred to as LnPz2 (Pz = porphyrazine), is shown in Figure 2. (Cartesian coordinates are given in the Supplementary Table S1). The same geometry was used for each of the [LnPz2]0 considered. All calculations on [LnPz2]0 were done without employing Cholesky decomposition of the two-electron integrals. The RAS2 space consists again of the seven 4f orbitals plus the π-SOMO. Seven additional occupied π-MOs are included in RAS1 (having a1 + b1 + b2 + e1 + e3 symmetries) and four unoccupied π∗-MOs in RAS3 (having e1 + e3 symmetries). Up to two holes/particles in RAS1/RAS3 were allowed. This space of 12 active π-orbitals was chosen to correspond to the in and out of phase combinations of the 6 frontier orbitals predicted by a Hückel model of the sixteen membered inner ring C8N8 of Pz. These Hückel orbitals have pseudo angular momenta λ = ±3, ±4, ±5.
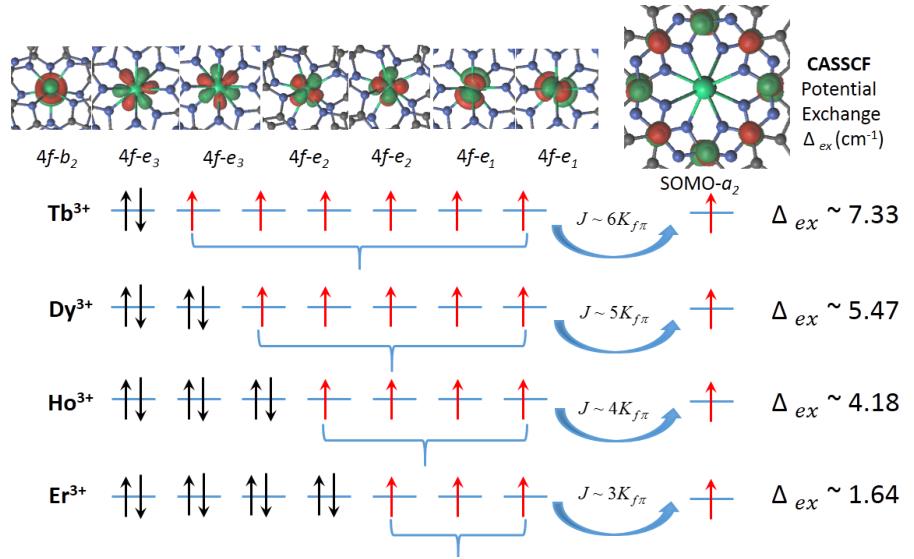 Figure 1. Schematic of the ferromagnetic exchange coupling mechanism in the CASSCF active space of [LnPc2]0. The active space consists of seven Ln 4f orbitals and the radical spin-1/2 orbital as shown on top, with their symmetry labels in the D4d point group. Kfπ represents a potential exchange integral between the π-SOMO and a 4f orbital, and J represents the total exchange strength.
Figure 1. Schematic of the ferromagnetic exchange coupling mechanism in the CASSCF active space of [LnPc2]0. The active space consists of seven Ln 4f orbitals and the radical spin-1/2 orbital as shown on top, with their symmetry labels in the D4d point group. Kfπ represents a potential exchange integral between the π-SOMO and a 4f orbital, and J represents the total exchange strength.
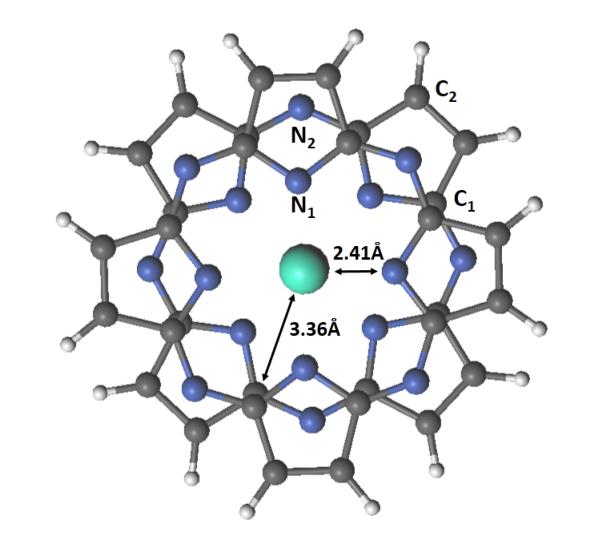 Figure 2. Molecular geometry of [LnPz2]0 used in the RASSCF calculations, where carbon (gray) and nitrogen (blue) atoms are classifed into four groups: C1, C2, N1 and N2. The Ln–N1 and Ln–C1 distances are also shown.
Figure 2. Molecular geometry of [LnPz2]0 used in the RASSCF calculations, where carbon (gray) and nitrogen (blue) atoms are classifed into four groups: C1, C2, N1 and N2. The Ln–N1 and Ln–C1 distances are also shown.
We found that the orbitals of e1 and e3 symmetry had a tendency to rotate out of the active space. To prevent this from happening, the 8 orbitals of e1 and e3 symmetry were put into an artificial symmetry class so as to disable orbital mixing with orbitals outside this class (using the “supersymmetry” keyword of MOLCAS). The validity of this approach relies on the quality of the starting orbitals. These were obtained from a state-averaged RASSCF calculation on the twofold degenerate ferromagnetic (S = 7/2) ground state of [TbPz2]0. This calculation did not experience the unwanted rotations and provided correct orbitals.
Before spin–orbit coupling (SOC) is considered, the exchange coupling between the lanthanide and the spin-1/2 radical can be evaluated as the energy difference between the high-spin and low-spin states that are obtained by coupling the total spin of the Hund term of the Ln3+ ion (7F for Tb3+, 6H for Dy3+, 5I for Ho3+, and 4I for Er3+) with the spin-1/2 of the radical. In each case, the state-averaging was performed over all states formally arising from the Hund term. As an example, for the [DyPc2]0 molecule, we optimize respectively S = 3 high-spin and S = 2 low-spin, with the state average including all 11 spatial components of the L = 5 Hund term 6H of the Dy3+ ion. We then evaluate the exchange gap as the difference between the lowest S = 3 and S = 2 energies.
Finally, SOC is introduced by matrix diagonalization in the basis of all the optimized S = 2 and S = 3 CASSCF/RASSCF wavefunctions.
We note that a similar approach was used in a recent computational study of the exchange interaction in the dimer Ce2(COT)3 [30].
The calculated CASSCF active natural orbitals of [LnPc2]0 are shown in the top of Figure 1. The Ln 4f orbitals are quasi atomic, while the spin-1/2 radical (π-SOMO) is mainly localized and evenly distributed on the C1 atoms with nodes on the N atoms. The exchange gaps obtained from the CASSCF calculations (without SOC) are listed in Table 1. Our calculations predict ferromagnetic intramolecular exchange for all four [LnPc2]0 molecules.
The occurrence of ferromagnetic exchange interaction in the CASSCF calculations can be explained on the basis of a symmetry analysis in the approximate D4d point group of the molecule: The SOMO of Pc2 transforms as a2 (see top right of Figure 1), while the seven lanthanide 4f orbitals transform as b2 + e1 + e2 + e3. Thus, the magnetic orbital containing the Pc2 radical is orthogonal by symmetry to each of the magnetic orbitals of the lanthanide ion. Kinetic exchange between the magnetic orbitals is therefore forbidden and only the ferromagnetic potential exchange interaction is allowed [31,32].
 Table 1. CASSCF exchange gaps (cm–1) of [LnPc2]0 without SOC. Positive numbers signify ferromagnetic coupling.
Table 1. CASSCF exchange gaps (cm–1) of [LnPc2]0 without SOC. Positive numbers signify ferromagnetic coupling.
Table 1 further shows that the calculated exchange gap decreases from [TbPc2]0 to [ErPc2]0. This may be understood by considering the number of unpaired 4f electrons: In the ground Russell–Saunders term of the Ln3+ ions considered here, the number of unpaired 4f electrons decreases with increasing overall number of 4f electrons (see Figure 1). If we consider that each unpaired 4f electron contributes additively to the overall exchange, the latter is expected to decrease in magnitude in going from [TbPc2]0 to [ErPc2]0, as observed in Table 1. We note in this respect a recent experimental work in which the magnetic coupling between TbPc2 and a Ni(111) surface was also found to decrease along the series Tb–Ni > Dy–Ni > Er–Ni [17].
Table 2 lists the lowest lying energy levels of [LnPc2]0 obtained after diagonalization of the SOC in the CASSCF states. There is a clear separation in energy between the group of four lowest states and the next group of states. It is known that the crystal field in these compounds gives rise to a splitting of the atomic J ground state multiplet of Ln3+ into a set of crystal field levels of which the ground state is a doublet [33,34]. Exchange coupling of this doublet with the spin-1/2 of the radical electron gives rise to the four low-lying levels seen in Table 2. In the same way, coupling of higher crystal field levels with the radical results in the groups of higher lying states in Table 2.
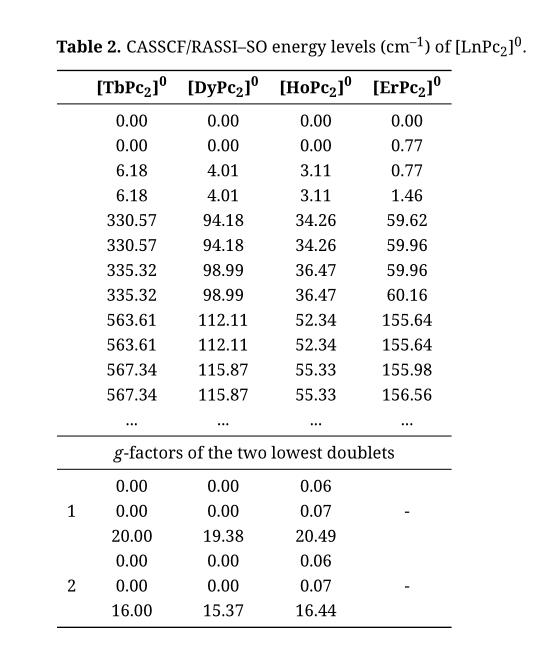 Table 2. CASSCF/RASSI–SO energy levels (cm–1) of [LnPc2]0.
Table 2. CASSCF/RASSI–SO energy levels (cm–1) of [LnPc2]0.
The total exchange splittings in the ground state are seen to be similar in magnitude to those before SOC, and are again found to decrease, from 6.18 cm–1 in [TbPc2]0 to 1.46 cm–1 in [ErPc2]0. The effective exchange coupling in the lowest doublet is of Ising type for Tb, Dy, Ho, but of anisotropic Heisenberg type for Er. We can attribute this difference in behavior to the different nature of the ground state doublet: [ErPc2]0 has MJ = ±1/2 as its ground doublet while [TbPc2]0, [DyPc2]0, [HoPc2]0 all have |MJ| > 1/2 ground states [33,34]. If we assume that the exchange between real spins is given by –2JSLn·SPc2 , projection on any doublet with |MJ| > 1/2 gives an effective Ising coupling: –2Jeff SLnz SPc2z, while projection on the MJ = ±1/2 doublet of Er gives an effective anisotropic Heisenberg coupling: –2Jeff(SLnz SPc2z + 8SLnx SPc2x + 8SLny SPc2y), where S Ln is the effective spin of the Ln doublet. Diagonalization of this anisotropic Heisenberg Hamiltonian gives a spectrum consisting of a nondegenerate ground state, followed by a doublet at 8|Jeff| – Jeff, and another nondegenerate state at 16|Jeff|. Referring to Table 2, we observe a qualitative agreement with the four lowest exchange states of [ErPc2]0.
Establishing the sign of the exchange interaction is not as straightforward for the calculations with SOC as it is for the calculations without SOC. For the cases where the exchange is of Ising type (Tb, Dy, Ho) it can be done by comparing the calculated g-factors of the lowest two doublets (see Tables 2 and 3). We see that the principal g-factors of the ground doublet are exactly 4 units higher than those of the next doublet. This corresponds to a spin flip of the radical electron (whose g-factor is 2), from ferromagnetic alignment in the ground doublet to antiferromagnetic alignment in the next doublet. Hence the coupling can be described as ferromagnetic. This straightforward interpretation cannot be applied to the case of Er however, because the exchange is not of Ising type there.
We now consider the effect of introducing π–π∗ correlation using the RASSCF method. The calculated values of the exchange gaps before SOC are given in Table 4. These calculations were done on the smaller model compounds [LnPz2]0. The absence of the outer benzene rings will affect the calculated spin density distribution and exchange coupling to some extent, but we expect that the physics of the exchange mechanisms will be correctly represented by the LnPz2 models. This is partly confirmed by comparing the CASSCF values of the exchange gaps in Tables 1 and 4: the sign, order of magnitude and trend are the same. Further confirmation is provided by the energy levels after SOC, which compare well between the LnPz2 (Supplementary Table S2) and LnPc2 (Table 2) complexes. Additional computational evidence for the relatively small influence of structural changes on the low-energy electronic structure of this family of molecules was provided by Pederson et al. [26].
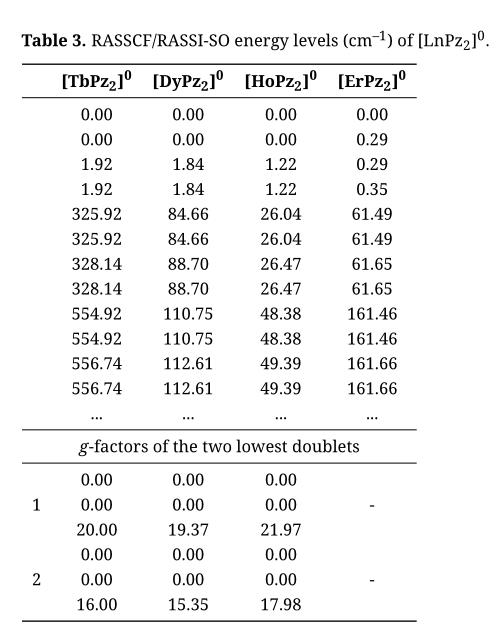 Table 3. RASSCF/RASSI-SO energy levels (cm–1) of [LnPz2]0.
Table 3. RASSCF/RASSI-SO energy levels (cm–1) of [LnPz2]0.
 Table 4. CASSCF and RASSCF exchange gaps (cm–1) of [LnPz2]0 without SOC. Positive numbers signify
ferromagnetic coupling.
Table 4. CASSCF and RASSCF exchange gaps (cm–1) of [LnPz2]0 without SOC. Positive numbers signify
ferromagnetic coupling.
The results in Table 4 show that the RASSCF exchange gaps are still ferromagnetic but smaller than the corresponding CASSCF gaps. This we interpret as the result of a competition between a new antiferromagnetic exchange pathway, opened up by activation of π–π∗ correlation, and the direct ferromagnetic exchange pathway already present in the CASSCF calculations.
The spin–orbit coupled RASSCF spectrum is given in Table 3. The exchange splittings are smaller than the corresponding CASSCF values (Table 2 and Supplementary Table S2) in line with the reduction of the SOC-free exchange splittings. We note in particular the value for Tb, which decreases from 6.18 cm–1 to 1.92 cm–1, closer to the experimental value of 0.88 cm–1 [23].
Note that the recent CASSCF calculations by Pederson et al. found Jeff = 8.2 cm–1 and 6.6 cm–1 for two geometries of [TbPc2]0, which is basically the same result we obtain with our CASSCF calculation, using an active space where 4f orbitals and the Pc2 SOMO only are considered. This seems to suggest that the π–π∗ excitations introduce by Pederson et al. in their active space were not sufficient to describe the spin-polarization antiferromagnetic exchange mechanism discovered in this work.
Interestingly, in the Er compound, the relative energies of the four lowest exchange states cannot be reproduced by a Hamiltonian of the form 2Jeff(SLnz SPc2z + 8SLnx SPc2x + 8SLny SPc2y), thus pointing to a likely breakdown of the 2JSLn · SPc2 approximation, a conclusion also reached in some recent experimental work on Ln–radical exchange interaction [35,36]. Recently, Chibotaru, Iwahara, et al. have discussed this breakdown on theoretical grounds using a microscopic model of exchange interaction [37–40]. Their model did not include spin polarization effects on the radical ligand and would thus have to be extended to be applicable to our case.
It should be noted that an antiferromagnetic exchange coupling pathway, as introduced in the RASSCF calculations, cannot be explained in terms of interaction between magnetic orbitals on the spin carriers. We have seen that the SOMO of Pc2/Pz2, belonging to the a2 irrep of D4d, is orthogonal by symmetry to the 4f orbitals of Ln3+. This absence of orbital overlap leads to stabilization of the high-spin state, i.e., ferromagnetic coupling [32,41].
We attribute this breakdown of the usual model to the effect of spin polarization in the π system of the Pc2/Pz2 radical. Spin polarization in radicals of conjugated π systems is a well known effect, and was invoked by McConnell to explain ferromagnetic coupling between stacked organic radicals (”McConnell’s first model”) [42]. Later, Yoshizawa and Ho mann argued that these magnetic couplings can be equally well explained on the basis of interaction between the SOMO’s of the organic radicals [41], the condition for ferromagnetic coupling being again the (near) vanishing of orbital overlap.
Let us now consider the spin density distribution in the Pc2/Pz2 radical. The SOMO (pictured in Figure 1) has amplitudes on the C atoms but nodes on all the N atoms. The spin density, in the simple molecular orbital picture, is therefore positive on the carbons but zero on the nitrogens. When we allow for electron correlation in the π system (as in our RASSCF calculations), small but negative spin densities appear on the N atoms. This is illustrated numerically with Mulliken spin populations in Table 5.
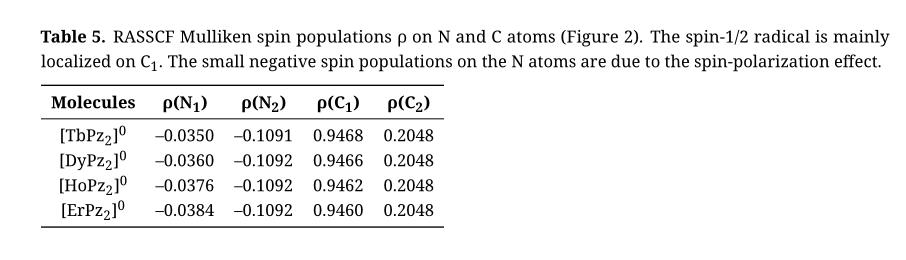 Table 5. RASSCF Mulliken spin populations ρ on N and C atoms (Figure 2). The spin-1/2 radical is mainly
localized on C1. The small negative spin populations on the N atoms are due to the spin-polarization effect.
Table 5. RASSCF Mulliken spin populations ρ on N and C atoms (Figure 2). The spin-1/2 radical is mainly
localized on C1. The small negative spin populations on the N atoms are due to the spin-polarization effect.
An elaborate analysis of the interplay between spin polarization and exchange in [LnPc2]0 will not be attempted here. Instead, a simple argument in the spirit of McConnell’s first model will be given. Let us assume then, that the total exchange splitting can be estimated as the sum of contributions from each atom of the ligand, and that only those atoms whose spin populations are non-zero can contribute. We can also assume that atoms further away from the central lanthanide ion will have a smaller exchange interaction with it than atoms closer by. Referring to Figure 2, the atoms closest to Ln3+ are the 8 N1 atoms at 2.41 Å and the 16 C1 atoms at 3.36 Å.
In the absence of spin polarization (the CASSCF case) there is only spin density on C1. Since all C1 atoms are symmetry related, the contribution from each of them to the exchange interaction must be the same. And since the overall interaction is ferromagnetic, the contribution from each C1 atom must be ferromagnetic as well. On the other hand, when spin polarization is allowed (the RASSCF case), the N1 atoms carry negative spin density, which will also interact with the lanthanide spin. If we may assume that this interaction is ferromagnetic, just like that of the C1 atoms, a competition arises: On the one hand, the majority spin on C1 atoms tries to align itself parallel to the Ln3+ spin, favoring overall ferromagnetic coupling. On the other hand, the polarized minority spin density on N1 atoms, with an opposite sign of spin compared with C1 atoms, also tries to be parallel to the metal spin, favoring overall antiferromagnetic coupling. As a result, the total exchange interaction is a sum of a positive contribution from C1 and a negative contribution from N1. Apart from the sign, it is not possible to determine a priori how large the contribution from N1 is compared to that from C1. This can be seen from considering the two parameters that will determine the size of the contribution: the spin density on the atom and the distance from the atom to the lanthanide ion. The spin density on N1 is smaller than on C1, but N1 is closer to the lanthanide than C1 (2.41 Å vs. 3.36 Å), so the exchange interaction due to spin density on N1 is stronger than that due to a same amount of spin density on C1. The resulting contribution from N1 can thus be smaller or larger in absolute value than the contribution from C1. If it is smaller, the overall exchange interaction is still ferromagnetic, but weaker than it was before spin polarization. On the other hand, if it is larger, the overall exchange interaction turns from ferromagnetic into antiferromagnetic. In our RASSCF calculations we observe the first case.
We have presented results of a computational study of the intramolecular exchange coupling between Ln3+ 4f electrons and the Pc2 radical in [LnPc2]0 (Ln = Tb, Dy, Ho, and Er) molecules. We performed a series of state-averaged CASSCF and RASSCF calculations with and without SOC. When SOC is not considered, CASSCF calculations with minimum active space show that the coupling between lanthanides and the radical are all ferromagnetic, and that the magnitude of the exchange gap drops as the central metal goes from Tb to Er. On the other hand, inclusion of additional π–π∗ excitations via RASSCF calculations suggests a key role played by the polarized spin density on the nitrogen atoms, induced by the spin polarization effect on the Pc2 radical. The negative spin density on the nitrogen atoms introduces an antiferromagnetic exchange pathway, weakening the overall ferromagnetic coupling strength between lanthanides and the radical.
The small ferromagnetic coupling calculated for [TbPc2]0 agrees with the latest experimental EPR evidence [23] but conflicts with the susceptibility measurements of Trojan et al. [19,20]. Their data could only be explained by a large antiferromagnetic coupling, which our calculations do not support.
AS and WVdH designed the study. HH performed the ab initio calculations with help from WVdH and AS. HH, WVdH and AS analysed the data and wrote the manuscript.
The authors declare that they have no conflicts of interest.
AS acknowledges support from the Australian Research Council (Future Fellowship no. FT180100519).
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
Huang H, Van den Heuvel W, Soncini A. Lanthanide-Radical Magnetic Coupling in [LnPc2]0: Competing Exchange Mechanisms Captured via Ab Initio Multi-Reference Calculations. Quantum Mater Res. 2020;1:e200003. https://doi.org/10.20900/qmr20200003

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




