Location:Home >> Detail
Med One 2016; 1: e160025. https://doi.org/10.20900/mo.20160025

1 Department of Cardiology, Xiangya Hospital of Central South University, Changsha, Hunan 410008, P.R. China
2 Department of Respiratory & Critical Care Medicine, Xiangya Hospital, Central South University, Changsha, Hunan 410008, P.R. China
3 Department of Physiotheray, Melbourne School of Health Sciences, The University of Melbourne, Melbourne, Victoria 3053, Australia
4 State Key Laboratory of Cardiovascular Disease, Fu Wai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, P.R. China
*Correspondence: Zaixin Yu, MD, PhD, FESC, Department of Cardiology, Xiangya Hospital of Central South University, No. 87, Xiangya Road, Changsha, Hunan 410008, P.R. China. Tel: +86-731- 84327492.
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant inherited disorder characterized by mucocutaneous telangiectases and arteriovenous malformations (AVMs) predominantly in the liver, lung and brain. Pulmonary hypertension (PH) is a devastating disease of the pulmonary vasculature, which could lead to an increase in pulmonary vascular resistance and eventually cause right heart failure or death. Early epidemiological studies suggested that PH is a rare complication of HHT, however, accumulated evidences have showed that the prevalence of PH in HHT is evaluated up to 74 %. Post-capillary PH is the most common type of PH in HHT, which most often results from hepatic arteriovenous malformations due to a high cardiac output state. Another kind of HHT is less frequent and associated with genetic mutations in ENG or ACVRL1, those patients are more likely to develope into precapillary pulmonary arterial hypertension. PH is associated with a higher mortality rate and worse prognosis, which largely affects the overall life-expectation among those HHT patients. However, ENG and ACVRL1 mutations can only explain the onset of HHT and arteriovenous malformations, revealing the precise mechanisms of the co-occurrence of both diseases deserve further scientific attentions. Our review aims to illustrate the epidemiology, clinical features, molecular biological basis, pathological characteristics and potential treatment options for these two close associated but seemingly “contradictory” diseases.
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant inherited disorder characterized by mucocutaneous telangiectases and arteriovenous malformations (AVMs) predominantly in the liver, lung and brain [1, 2]. Generally, spontaneously and recurrently epistaxis is always the first and earilist symptom, which happens frequently from childhood [1]. The clinical diagnosis is based on the Curaçao criteria [3](Table 1), established by the HHT Foundation International, are useful for clinical diagnosis.
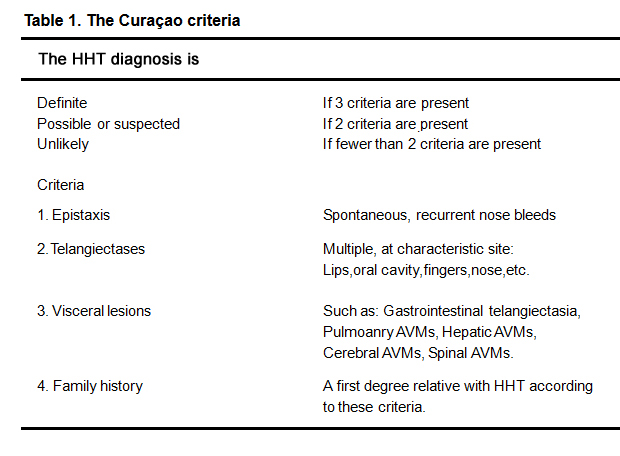 Table 1. The Curaçao criteria
Table 1. The Curaçao criteria
Pulmonary hypertension (PH) is a devastating disease of the pulmonary vasculature in which vasoconstriction and vascular remodeling both lead to a progressive increase in pulmonary vascular resistance (PVR). The right ventricular (RV) adaptation to chronic pressure overload would cause shortness of breath, dizziness, fainting, leg swelling and other symptoms, then eventually result in right heart failure [4, 5]. PH is a haemodynamic condition assessed by right heart catheterisation (RHC) with mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg [4, 5]. According to the most recent classification, PH can be one of six different groups (Table 2) [4, 5]. There are two main groups of PH can be divided according to the origin, the pre- and post capillary PH [4, 5]. Echocardiography is considered to be the foundation of the initial diagnosis of PH patients [4, 6].
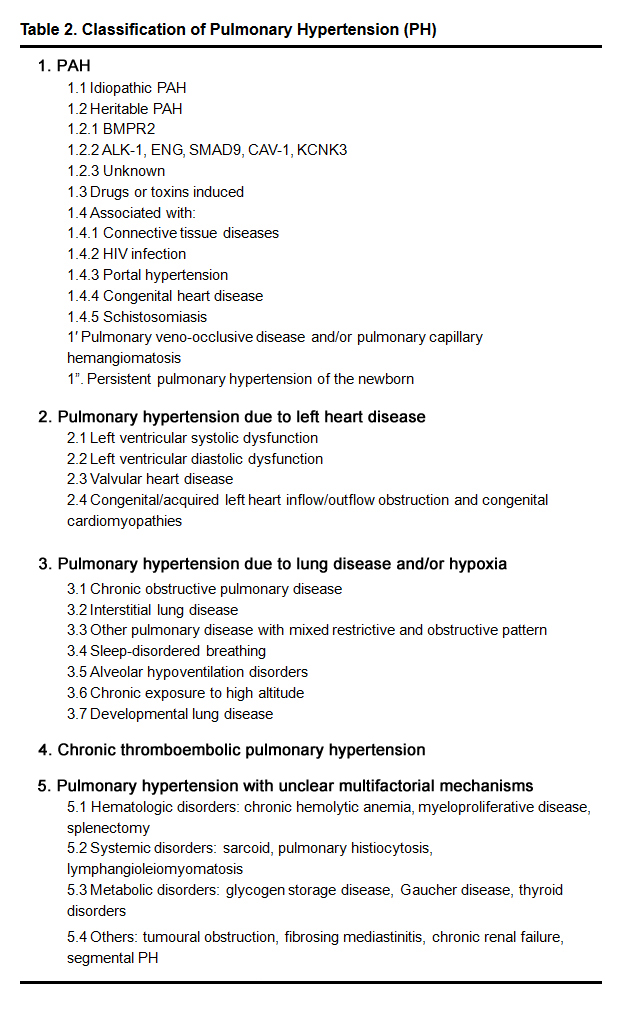 Table 2. Classification of Pulmonary Hypertension (PH)
Table 2. Classification of Pulmonary Hypertension (PH)
PAH, pulmonary arterial hypertension; PH,pulmonary hypertension; ALK-1, activin receptor-like kinase 1; BMPR2, bone morphogeneticotein receptor 2; CAV-1, caveolin-1; ENG, endoglin; KCNK3, potassiumhannel subfamily K member 3; SMAD9, mothers against decapentaplegic 9. Our review aims to clarify the epidemiological, pathophysiological, molecular genetics and therapeutic connections between these two seemingly “controversial” diseases.
While scarce data was available in early epidemiological studies on the prevalence of PH in patients with HHT, most authors agree that it is a rare complication, found in less than 1 % of patients with HHT [7]. However, with the intensive study of pathophysiologic mechanisms behind these two diseases and advanced diagnosis methods, the prevalence of PH in the HHT patients is evaluated up to 74 % [8-11].
Olivieri et al. [8] first looked on sixty-eight patients diagnosed with HHTand identified nine of them complicated with pulmonary hypertension by echocardiography, which is accounted for 13.2 %(9/68) of all patients . Among those nine patients diagnosed with both diseases, five of them were found with hepatic arteriovenous malformation (HAVMs) [8]. Subsequently, Sopeña and his colleague [9] found a high prevalence of possible or probable PH by echocardiography. PH was found in 29 hospitalized patients with HHT, which is accounted for 31 % (13/29) [9]. The PH was diagnosed by echocardiography with a mean right ventricular systolic pressure (RVSP) of over 56mmHg. Only three patients were further examined by right cardiac catheterization (RHC) and verified the PH diagnosis [9]. The study also found that ten out of those thirteen PH patients were diagnosed with arteriovenous malformation (83 %),and HAVM was the most common one which is documented in 67 % of these patients. By contrast, among these 29 patients who were not combined with PH, only 13 % of them were diagnosed with arteriovenous malformation. Thus, we could see that HHT complicated with PH is certainly more frequent than previously thought. Moreover, this study indicated that PH is leading to a poorer prognosis among HHT patients. During the follow-up, 19 patients passed away at an age of 73 ± 8.1, among those patients, ten patients died before 75 years old, and seven out of ten of them were previously diagnosed with PH. Eight HHT patients who were not complicated with PH all had a longer life expectation [9]. This finding indicates that PH is not only one of common complications of HHT, but also largely affects the final prognosis and life expectation of a HHT patient. But echocardiography is not the gold standard in the diagnosis of pulmonary hypertension, more often, it could only provide an evidence of the possibility of PH.
Another study by Lyle et al. [10] illustrated that 74 % of HHT patients who underwent RHC and echocardiography were diagnosed PH. The mean pulmonary arterial pressure (MPAP) of these 28 patients is fluctuating at 41 ± 11 mmHg, pulmonary arterial wedge pressure (PAWP) is at 17±11mmHg [10]. Of those 28 patients, twelve of them were diagnosed with pulmonary arterial hypertension by RHC [10]. Two of them were diagnosed with pulmonary hypertension secondary to high cardiac output or atrial septal with a normal PAWP and PVR [10]. Fourteen patients (50 %) had elevated PAWP ( ≥ 15 mm Hg), nine with evidence of high flow. During the follow-up, thirteen patients died, twelve out of thirteen patients were previously diagnosed with PH. The life expectation of HHT is dramatically dropped when is complicated with PH. The average lifespan is about 48 years, but among the patients with PH, their lifespans shorten 7 years [10]. This study also indicated that HHT patients were more likely to have a worse life expectation in HHT patients who associated with PH despite the causes. A newest study by Buonamico et al. [11] showed that among 153 HHT patients who were screened for HAVMs by abdominal multislice CT (MSCT), biological tests, and echo-color-Doppler [11], CT finding was positive in 84 % patients while Doppler color spots were also found in 86 % patients [11]. As we have mentioned above, the prevalence rate of HHT patients complicated with PH is fluctuated from 1 % to 74 %. Thus, we conclude that not all HHT patients who have evidences of HAVMs would develop into PH, also, not all HHT patients who have been diagnosed with PH would find the evidences of HAVMS. Among those HHT patients who were complicated with PH, but without any evidences of HAVM, the pathogenesis of the occurrence of PH remains unclear. Studies have showed that this kind of pulmonary hypertension is always along with ENG or ACVRL1 gene mutations and the pathological changes are quite similar to idiopathic pulmonary arterial hypertension [12, 13].
2.2 Pathophysiology of PH in HHTTelangiectasias and arteriovenous malformations are two main pathological changes of HHT, which are thought to be caused by changes in angiogenesis. While the disease progressively aggravating, postcapillary venule and its bifurcates intertwine and form abnormal vascular structures, postcapillary venules are lack of elastic fiber layer and meanwhile the vascular smooth muscle cell proliferation, postcapillary venules and precapillary arterioles link together and form arteriovenous shunts [14, 15]. From the pathophysiological perspective, HHT and PH are completely different and even controversial diseases. However, as aforementioned, pulmonary hypertension is more and more regarded as a vital complication of HHT, which indicate that HHT and PH share certain mechanisms during the diseases progress.
Several mechanisms underlie HHT induced PH. Most frequently post-capillary PH may result form a heart failure associated with high cardiac output (CO) due to HAVMs [16]. When the hepatic AVMs occur, the shunting of blood directly from hepatic artery to hepatic vein can result in a elevation in hepatic blood flow and an enlargement of circulatory volume [17, 18]. Systemic vasodilation could cause a drop in peripheral vascular resistance, then lead to a decrease in PCWP and blood pressure, at this period of time, the patients usually have symptoms of collapsing pulse. Then, in order to compensate to low blood pressure, the body automatically adjusts to a higher cardiac output and heart rate. At this period of time, the patients are suffered a decreased pulse pressure and sinus tachycardia [19]. The increase of cardiac output can enlarge the pulmonary blood flow and the pulmonary arterial pressure, which finally lead to pulmonary arterial hypertension [15]. We can see from the RHC characteristics of high-output PH with a gently elevation in mPAP, a normal PVR, increased PAWP and most significantly, a rise in CO [6].
Another kind of pulmonary hypertension associated with HHT is recognized as precapillary PH, which is relatively rare [13]. Patients with a diagnosis of this type of PH should systematically screen for underlying mutations of BMPR2 gene (bone morphogenetic protein receptor type 2), ACVRL1 (activine receptor-like kinase type 1), ENG (endogline) or Smad8 genes. The precapillary PH is similar to idiopathic PAH, which is characterized by remodeling of small pulmonary arteries and increasing of pulmonary vascular resistance [20]. In this kind of PAH, on the other hand, the RHC characteristics are differ from the high-output PH that the mPAP, PVR are usually higher due to pulmonary arteries remodeling, most importantly, with a normal or decreased CO and PAWP [20].
Since the treatments strategies towards postcapillary high output PH or pre-capillary PH were different, high-flow state with PH is usually treated with salt restriction and diuretics while pre-capillary PH might have to be treated by vascular-targeted therapy [6]. It is highly recommended that all HHT patients should finish echocardiography. We should always remember that echocardiography is not the gold standard in the diagnosis of pulmonary hypertension, more often, it could only provide a evidence of the possibility of PH. For the reason that treatments toward these two types of PH are totally different, it is essential to adopt RHC to clarify the hemodynamics among those HHT patients who have already had a high echocardiographic suspicious of PH.
2.3 Genetics and Molecular Biology of PH in HHT 2.3.1 Genetics and pathogenesis in HHTHHT is one of the most common kinds of autosomal dominant vascular dysplasia diseases that affect 1 in 10,000 individuals [19-22]. There are five genes are currently recognized as the disease-causing mutations. Of these, three have been linked to particular genes, while the two remaining have currently only been associated with a particular locus [21]. The majority of HHT patients are type HHT1 or HHT2, which were found with mutations in ENG encoding endoglin [22] or ACVRL1 encoding activin receptor-like kinase (ALK1),respectively [23]. A third genetic type is recognized as the SMAD4 gene, whose mutations also cause the gastrointestinal epithelial precancerous state of juvenile polyposis [24]. Another two unidentified genetic mutationsare associated with pure HHT, HHT3 between 141.9 and 146.4 Mb on chromosome 5q [25], and HHT4 on chromosome 7p between D7S2252 and D7S510 [26].
Human Endoglin (CD105) is a glycoprotein with a molecular weight of 180kDa, which is found on endothelial cells of capillaries, veins, and arteries [22, 27]. Murine endoglin is highly expressed on all endothelial cells and part of mesenchymal stromal cells [28]. Endoglin is constituted of 653 amino acid residues and regarded as relatively endothelial specific co-receptor complexes among the TGF-ß superfamily [28]. Considering the multiple functions of endoglin during the course of HHT [29], endoglin has a direct interaction with endothelial-specific type I receptor, ALK-1 [30], which is further structurally and mechanistically associated with BMP branch of type I receptors [31], type II receptors,BMPR2,and TβRII. Endoglin mainly activates the TGFβ-ALK1 signaling pathway and suppresses TGFβ-ALK5 signaling pathway [32] (Fig. 1).
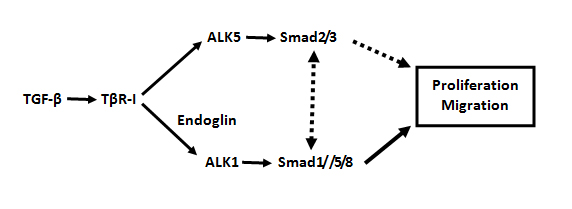 Fig. 1 TGF-β can regulateSmad signaling pathway with opposite effects in ECs.
Fig. 1 TGF-β can regulateSmad signaling pathway with opposite effects in ECs.
The TGF-β/ALK5 signaling pathway inhibits cell proliferation and migration, while the the TGF-β/ALK1 pathway promotes ECs proliferation and migration.
ACVRL1 (activin receptor-like kinase 1, or ALK1) gene encodes type I cell-surface receptor on the TGF β signaling pathway, which is consist of a glycine- and serine-rich region (GS domain), serine-threonine kinase sub-domains, and a short C-terminal tail [36]. Mutations in ACVRL1 are responsible for HHT2, a disease manifesting as fragile vessels, capillary overgrowth, and numerous AVMs [37]. In HHT patients, the majority of vessels are normal, however the ACVRL1 mutations could lead to abnormal angiogenetic responses and formation of anormalous arteriovenous connections, ranging from mucomembranous telangiectases to large arteriovenous malformations that can occur in every organ such as lungs, liver and brain [12, 38]. ENG or ACVRL1 mutations were found in more than 80 % of all HHT patients. One to two percent of cases 127 have mutations in SMAD4 [39]. This gene encodes smad proteins which is a member of signal transduction proteins in Smad family. The proteins are phosphorylated by transmembrane kinases and activated in response to TGF-β signaling pathway. Mutations or deletions in SMAD4 gene have been found in juvenile polyposis syndrome, pancreatic cancer, and HHT [40].
Large scale studies have not been presented, but it is widely accepted that most HHT arises from ENG or ACVRL1 haploinsufficiency, leading to the fact that the remaining wild type allele cannot contribute sufficient and effective protein to main a normal function.
2.3.2 Genetics and pathogenesis in PHPulmonary hypertension is a deadly disease in which vasoconstriction and vascular remodeling both lead to a progressive increase in pulmonary vascular resistance. It has been well studied that BMPR2 mutations are related to primary pulmonary arterial hypertension [6]. BMPR2 gene encodes transmembrane serine/threonine kinases in the bone morphogenetic protein (BMP) receptor family [41] and BMPs are members of the TGF-β signaling superfamily. It is widely accepted that BMPR2 could inhibit vascular smooth muscle tissue proliferation and prolong the viability of pulmonary arterial endothelial cells, while at the same time prevent arterial remodeling and alleviate inflammation [41]. The BMPR2 mutations result in a increase in pulmonary vascular resistance and pulmonary vascular hypertrophy , which eventually could lead to pulmonary hypertension and progressively right ventricular remodeling [42]. BMPR2 mutations were found in more than 70 % of familial cases, but were not found in any HHT associated PH [43]. Besides BMPR2 disease-causing mutations, subsequently, in some PH patients, ALK1、Endoglin and SMAD8 gene mutations on TGF-β pathway have also been found.
Studies have showed that PH patients with early onset and severe symptoms were usually found carrying a BMPR2 mutation. Those patients were less likely to respond to acute vasodilator testing [44-47]. But studies on the association between the PH phenotypes and the ACVRL1 mutations were quite rare. In Girerd et al. study [38, 39], 379 idiopathic or heritable PH patients were provided with BMPR2 and ACVRL1 genetic counseling and screening. The results showed that 277 patients were noncarriers, 93 found with BMPR2 mutations and 9 found with ACVRL1 mutations [38]. Those with ACVRL1 mutations are more likely to diagnosis PH at a younger age when compared to BMPR2 mutation carriers or noncarriers. The results showed that the majority of PH patients carrying ACVRL1 mutations were found to be children or teenagers [38]. It is also found that PH patients with ACVRL1 mutations were tend to suffer a poorer prognosis when compared to those BMPR2 mutation carriers, even though those ACVRL1 mutation carriers usually had a better initial hemodynamic status. It seems like ACVRL1 mutations could aggravate disease progress to a certain degree even with proper PH-target therapy. The study further provided evidences that PH could be the first onset symptoms of ACVRL1 mutation carriers [38]. Unfortunately, this study didn’t have any data on PH carrying ENG mutations.
As we have mentioned above, the majority of HHT occur due to the Endoglin or ACVRL1 [23]. Interestingly, both Endoglin and ACVRL1 genes encode cell surface protein receptors on the TGF-β/BMP signaling pathways. We conclude that the molecular pathogenesis of both diseases is related to gene mutations on the TGF-β signaling pathway.
2.3.3 Genetics and phenotypes connections of PH in HHTHHT is an autosomal dominant disease with a late onset and 97 % complete penetrance by the age of sixty years [48, 49]. In 2001, ACVRL1 mutations were found in the developemtn of PH for the first time. After, it was further confirmed by finding ACVRL1 mutations in other PH patients along with clinical symptoms of HHT [6, 12, 13]. Also, PH associated with HHT is due to a dysfunction of ALK-1 protein [50]. Thus, it is highly suggested that there is a link between the two totally different diseases of PH and HHT concerning ACVRL1 mutations. Less frequently, PH with endoglin mutation patients have also been reported, which drive the attention to the association between PH and HHT [38].
However, BMPR2 mutations were detected in approximately 70 % of hereditable PH and 15-25 % idiopathic PH patients, but were not found in any HHT associated PH [38]. Rigelsky et al. [51] reported a 36 years old woman initially diagnosed with PH at the age of 24. And at her 35, this patient is discovered with multiple pulmonary arteriovenous malformations and history of epistaxis [51]. Interestingly, mutations detecting of three HHT disease-causing genes, ACVRL1, ENG, and SMAD4, were all normal. However,a nonsense mutation were then identified in BMPR2 gene [51]. Unfortunally, the patient didn’t have any family histories of HHT, also there were some disagreements on the clinic diagnosis of nasal telangiectases and repeated epistaxis. According to the Curaçao diagnostic criteria [3] of HHT, this patient only could be diagnosed as suspected HHT. Still, there is a possibility that this patient may become the first known report of a HHT patient caused by a BMPR2 mutation. This case highlighted the importance of BMPR2 gene detection in patients associated with both HHT and PH.
The heterogeneity of HHT represents many differences in age onset and the severity of clinical symptoms [52], which has been partially explained by the identification of ACVRL1 and ENG genes mutations. Studies have showed that ACVRL1 mutations could result in both capillary vascular dilation and pulmonary arteries hypertrophy simultaneously [20]. Accumulated evidences indicate that symptoms and features are similar in both types of HHT patients, but the occurrence of specific arteriovenous malformations varies greatly with the different genotype. It is evident that more HHT1 patients were found with cerebral AVMsthan those HHT2 patients. For instance, studies have showed that intrapulmonary shunting was found in 85 % of typer 1 HHT patients, 35 % of type 2 HHT patients [53],and compared to 7 % of normal controls. Juvenile polyposis (JP) only found in patients with SMAD4 mutations. Type 2 HHT patients have a higher prevalence rate of HAVMs [54-56], and more HAVMs-related post capillary pulmonary hypertension as well [57, 58]. Pulmonary arterial hypertension is also more common in HHT2 patients [8, 59, 60]. Due to the early development of PH and the late-onset of HHT manifestations, some patients with ACVRL1 mutations may develope a severe PH without any clinical evidences of HHT [38] .
Although increasing number of evidences have showed the probable and potential association in the TGF-β signaling pathway underlying the pathophysiology of both PH and HHT, but the precise mechanism is not fully understand.
2.4 Treatment of PH in HHTPH-target therapy may increase the bleeding complication of HHT, and the vasoconstrictive treatment for the telangiectasia may also exacerbate the pulmonary arterial stenosis. We could see that there is a certain contradiction for the treatment of PH and HHT. Although, there are many current studies have associated PH with HHT, no systematic evidence has showed any exact anti-PH therapeutic protocols for the overall HHT population.
The first-line treatment for high-output PH consists of an adequate control of bleeding episodes and maintain a normal hemoglobin level, salt restriction, diuretics, digoxin and other symptomatic supportive treatment if necessary [61]. When those patients are found resistant to classical medication therapy, liver transplantation could be the final option, especially for those hepatic arteriovenous malformations induced PH [62]. But the morbidity is still high [63, 64], therefore, it’s not recommended because of the disadvantages of the surgical morbidity, mortality, and an immunosuppressive treatment for life.
Is liver transplant the only treatment available for restoring normal cardiac output and reverse high-output PH in patients with HHT? The answer is no. A recent study has showed that Bevacizumab as an anti–vascular endothelial growth factor may be a beneficial treatment for HHT patients with severe HAVMs related high cardiac output [19]. Twenty-five HHT-related high cardiac output patients were treated with bevacizumab for up to 6-month follow-up period. Median cardiac index was significantly dropped from 5.05 L/min/m2 to 4.2 L/min/m2 after three months of bevacizumab treatment [19]. After another 3 months, the median cardiac index was also obviously lower than the initial stage of the treatment. While the mean duration of epistaxis was greatly decreasedat 3 months and 6 months respectively, the quality of life had a considerable improvement [19]. It is showed that bevacizumab treatment contributes to preventing a high cardiac output, which indicated that this may be an efficient medication for HHT associated with high-output PH.
With respect to the treatment of the pulmonary, there are three PH-target medications have been widely used in clinic: endothelin receptor antagonists (ERA), phosphodiesterase inhibitors (PD5I) and prostacyclins [6]. From 2006 to 2016, there were only three cases have described effective treatment of PH in HHT patients with PH-target medications and gave us a suggestion on choosing for a better medication [65-67]. Among these three patients, two were treated with well-known ERA bosentan [65, 66], and one was treated with PD5I sildenafil [67]. After the treatment, the dyspnea symptom and exercise capacity were ameliorated, laboratory examination was found a decrease in mPAP [65-67]. Those cases indicated that the PH-target therapy could also effectively improve the overall symptoms of PH in HHT patients. However, there was no report concerning other PH-target medication besides ERA and PD5I in patients with both diseases.
It is less recommended by using oral anticoagulation medication in HHT patients due to the increase danger of bleeding complication [21, 68]. However, in recent literature, anticoagulation therapy could be considered in some cases [68]. Lung transplantation is considered as remaining treatment for those patients who suffer from severe PH [45]. However, till now, there was no one did lung transplantation on a ACVRL1 mutation carrier partly due to the shortage of donor lungs, but more importantly because the evidence of arteriovenous malformations and associated high risk in those HHT patients [38].
It's a priority to treat patients with PH according to the guidelines for PH and meanwhile to give consideration to supporting-therapy such as diuretics, oxygen, and digoxin [6].
In summary, hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant inherited disorder characterized by mucocutaneous telangiectases and AVMs [1]. Pulmonary hypertension is increasingly recognized as a severe complication of HHT. There are close correlations and also unexplainable contradictions among these two diseases. With the advances in scientific and medical understanding of the pathophysiologic mechanism behind these two diseases, PH has a higher prevalence in HHT patients than previously thought. But data on the exact prevalence of PH in the overall HHT population was inadequate, partly because most studies only included a small sample size of patients, most importantly, due to the lack of RHC evidences to further prove the occurrence of PH. The majority of HHT-related PH was post-capillary, which was caused by HAVMs and anemia associated high cardiac output. Less frequently, ACRVL-1 or ENG mutations result in pre-capillary PH, which was clinically and histological similar to idiopathic pulmonary artery hypertension. According to previous studies, overall life expectations of HHT patents that develop an evidence of PH were much lower than those HHT patents alone. Therefore we first recommend a systematic screening to reveal the true prevalence of both forms of PH in a HHT population and then we reinforce the importance of RHC to clarify hemodynamic.
Accumulated evidences have showed that there is a probable and potential involvement of the TGF-β signaling pathway in the pathophysiology of both PH and HHT, but the precise mechanism is not fully understood. But neither of BMPR2, Endoglin or ACRVL-1 gene mutations is able to explain the co-occurrences of HHT and PH. Thus, we suggest a possibility that there are some modifier genes or small molecules on the TGF-β signaling pathway are involved in causing and modulating the mechanism of both diseases, or there may exist another undetected disease-causing mutation could explain the co-occurrence of both diseases. However, revealing the precise mechanisms and finding suitable therapeutic options behind these two diseases, deserve further scientific attentions.
This work was supported by the National Natural Science Foundation of China (program number: 81570050) and Innovation Project Foundation of Central South University (2016zzts530).
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
He J, Li Y, He J, Wang X, Yu Z. Associations Between Two Seemingly “Contradictory” Diseases: Hereditary Hemorrhagic Telangiectasia and Pulmonary Hypertension. Med One. 2016 Dec 25; 1: e160025. https://doi.org/10.20900/mo.20160025

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions



